Background
Three-dimensional (3-D) cultures of cancer cells can potentially bridge the gap between 2-D drug screening and in vivo xenografts. The objective of this study ... More
Background
Three-dimensional (3-D) cultures of cancer cells can potentially bridge the gap between 2-D drug screening and in vivo xenografts. The objective of this study was to characterize the cellular and extracellular matrix characteristics of spheroids composed of human lung epithelial cells (epi), pulmonary vascular endothelial (endo) cells, and human marrow-derived mesenchymal stems cells (MSCs).
Methods
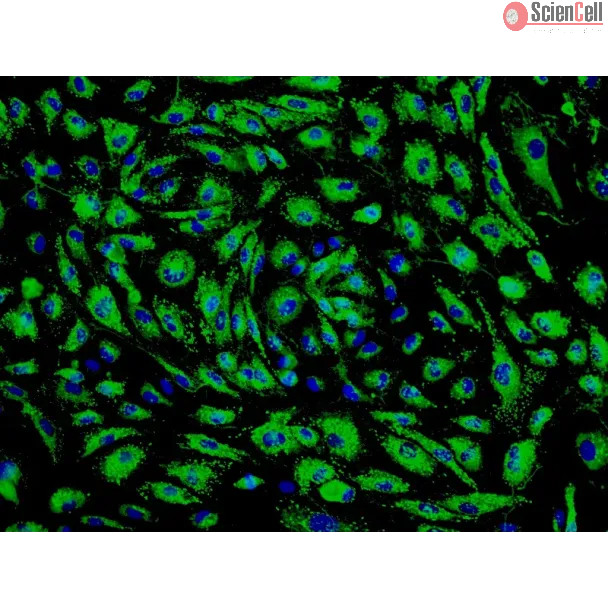
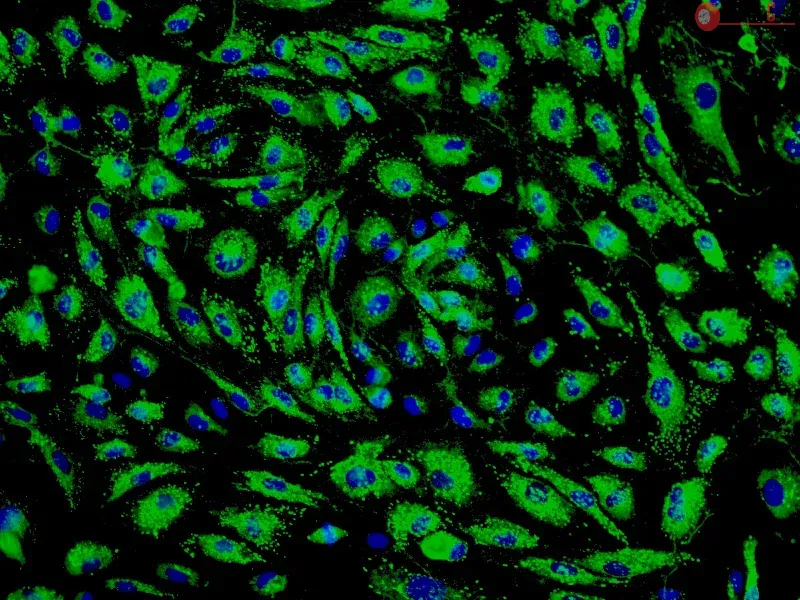
Spheroids composed of epi/endo/MSCs, termed herein as synthetic tumor microenvironment mimics (STEMs), were prepared by the hanging drop method. Cellular composition and distribution in the STEMs was characterized using fluorescence microscopy. Induction of reactive oxygen species and upregulation of efflux transporters was quantified using fluorometry and PCR, respectively, and phenotypic markers were qualitatively assessed using immunohistochemistry.
Results
STEMs exhibited three unique characteristics not captured in other spheroid cultures namely, the presence of a spheroid core devoid of epithelial cells and primarily composed of MSCs, a small viable population of endothelial cells hypothesized to be closely associated with MSCs within the hypoxic core, and discrete regions with high expression for vimentin and cytokeratin-18, whose co-expression is co-related with enhanced metastasis. Although cells within STEMs show elevated levels of reactive oxygen species and mRNA for ABC-B1, an efflux transporter associated with drug resistance, they exhibited only modest resistance to paclitaxel and gemcitabine in comparison to 2-D tri-cultures.
Conclusions
The epi/endo/MSC spheroid model described herein offers a promising platform for understanding tumor biology and drug testing in vitro. Less
Background: Mesenchymal stem cells (MSCs) stabilise endothelial barrier function in acute lung injury via paracrine hepatocyte growth factor (HGF). Vascular endothelial g... More
Background: Mesenchymal stem cells (MSCs) stabilise endothelial barrier function in acute lung injury via paracrine hepatocyte growth factor (HGF). Vascular endothelial growth factor (VEGF), which is secreted by MSCs, is another key regulator of endothelial permeability; however, its role in adjusting permeability remains controversial. In addition, whether an interaction occurs between HGF and VEGF, which are secreted by MSCs, is not completely understood. Methods: We introduced a co-cultured model of human pulmonary microvascular endothelial cells (HPMECs) and MSC conditioned medium (CM) collected from MSCs after 24 h of hypoxic culture. The presence of VEGF and HGF in the MSC-CM was neutralised by anti-VEGF and anti-HGF antibodies, respectively. To determine the roles and mechanisms of MSC-secreted HGF and VEGF, we employed recombinant humanised HGF and recombinant humanised VEGF to co-culture with HPMECs. Additionally, we employed the RhoA inhibitor C3 transferase and the Rac1 inhibitor NSC23766 to inhibit the activities of RhoA and Rac1 in HPMECs treated with MSC-CM or VEGF/HGF with the same dosage as in the MSC-CM. Then, endothelial paracellular and transcellular permeability was detected. VE-cadherin, occludin and caveolin-1 protein expression in HPMECs was measured by western blot. Adherens junction proteins, including F-actin and VE-cadherin, were detected by immunofluorescence. Results: MSC-CM treatment significantly decreased lipopolysaccharide-induced endothelial paracellular and transcellular permeability, which was significantly inhibited by pretreatment with HGF antibody or with both VEGF and HGF antibodies. Furthermore, MSC-CM treatment increased the expression of the endothelial intercellular adherence junction proteins VE-cadherin and occludin and decreased the expression of caveolin-1 protein. MSC-CM treatment also decreased endothelial apoptosis and induced endothelial cell proliferation; however, the effects of MSC-CM treatment were inhibited by pretreatment with HGF antibody or with both HGF and VEGF antibodies. Additionally, the effects of MSC-CM and VEGF/HGF on reducing endothelial paracellular and transcellular permeability were weakened when HPMECs were pretreated with the Rac1 inhibitor NSC23766. Conclusion: HGF secreted by MSCs protects the endothelial barrier function; however, VEGF secreted by MSCs may synergize with HGF to stabilise endothelial cell barrier function. Rac1 is the pathway by which MSC-secreted VEGF and HGF regulate endothelial permeability. Less
Small interfering RNA (siRNA) targeted therapeutics (STT) offers a compelling alternative to tradition medications for treatment of genetic diseases by providing a means ... More
Small interfering RNA (siRNA) targeted therapeutics (STT) offers a compelling alternative to tradition medications for treatment of genetic diseases by providing a means to silence the expression of specific aberrant proteins, through interference at the expression level. The perceived advantage of siRNA therapy is its ability to target, through synthetic antisense oligonucleotides, any part of the genome. Although STT provides a high level of specificity, it is also hindered by poor intracellular uptake, limited blood stability, high degradability and non-specific immune stimulation. Since serum proteins has been considered as useful vehicles for targeting tumors, in this study we investigated the effect of incorporation of human serum albumin (HSA) in branched polyethylenimine (bPEI)-siRNA polyplexes in their internalization in epithelial and endothelial cells. We observed that introduction of HSA preserves the capacity of bPEI to complex with siRNA and protect it against extracellular endonucleases, while affording significantly improved internalization and silencing efficiency, compared to bPEI-siRNA polyplexes in endothelial and metastatic breast cancer epithelial cells. Furthermore, the uptake of the HSA-bPEI-siRNA ternary polyplexes occurred primarily through a caveolae-mediated endocytosis, thus providing evidence for a clear role for HSA in polyplex internalization. These results provide further impetus to explore the role of serum proteins in delivery of siRNA. Less
Background: The mechanisms by which bacterial ligands alter angiogenesis remain unknown. Results: Lipopolysaccharide-mediated Angiopoietin-2-dependent autocrine angiogene... More
Background: The mechanisms by which bacterial ligands alter angiogenesis remain unknown. Results: Lipopolysaccharide-mediated Angiopoietin-2-dependent autocrine angiogenesis in lung endothelial cells is regulated by NADPH oxidase 2. Conclusion: Endothelial Nox2 regulates Angiopoietin-2-dependent angiogenesis. Significance: This study presents new data regarding the regulation of proinflammatory angiogenesis. Keywords: Angiogenesis, Endothelial Cell, LPS, Lung Injury, NADPH Oxidase, Angiopoeitin-2 Less
Autophagy, a type II programmed cell death, is essential for cell survival under stress, e.g. lung injury, and bone marrow-derived mesenchymal stem cells (BM-MSCs) have g... More
Autophagy, a type II programmed cell death, is essential for cell survival under stress, e.g. lung injury, and bone marrow-derived mesenchymal stem cells (BM-MSCs) have great potential for cell therapy. However, the mechanisms underlying the BM-MSC activation of autophagy to provide a therapeutic effect in ischaemia/reperfusion-induced lung injury (IRI) remain unclear. Thus, we investigate the activation of autophagy in IRI following transplantation with BM-MSCs. Seventy mice were pre-treated with BM-MSCs before they underwent lung IRI surgery in vivo. Human pulmonary micro-vascular endothelial cells (HPMVECs) were pre-conditioned with BM-MSCs by oxygen-glucose deprivation/reoxygenation (OGD) in vitro. Expression markers for autophagy and the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) signalling pathway were analysed. In IRI-treated mice, administration of BM-MSCs significantly attenuated lung injury and inflammation, and increased the level of autophagy. In OGD-treated HPMVECs, co-culture with BM-MSCs attenuated endothelial permeability by decreasing the level of cell death and enhanced autophagic activation. Moreover, administration of BM-MSCs decreased the level of PI3K class I and p-Akt while the expression of PI3K class III was increased. Finally, BM-MSCs-induced autophagic activity was prevented using the inhibitor LY294002. Administration of BM-MSCs attenuated lung injury by improving the autophagy level via the PI3K/Akt signalling pathway. These findings provide further understanding of the mechanisms related to BM-MSCs and will help to develop new cell-based therapeutic strategies in lung injury. Less
Carbon plasma nanocoatings with controlled fraction of sp3-C bonding were deposited on TiO2 nanorod arrays (TNAs) by DC magnetic-filtered cathodic vacuum arc deposition (... More
Carbon plasma nanocoatings with controlled fraction of sp3-C bonding were deposited on TiO2 nanorod arrays (TNAs) by DC magnetic-filtered cathodic vacuum arc deposition (FCVAD). The cytocompatibility of TNA/carbon nanocomposites was systematically investigated. Human umbilical vein endothelial cells (HUVECs) were cultured on the nanocomposites for 4, 24, and 72 h in vitro. It was found that plasma-treated TNAs exhibited excellent cell viability as compared to the untreated. Importantly, our results show that cellular responses positively correlate with the sp3-C content. The cells cultured on high sp3-C-contented substrates exhibit better attachment, shape configuration, and proliferation. These findings indicate that the nanocomposites with high sp3-C content possessed superior cytocompatibility. Notably, the nanocomposites Introduction: Mesenchymal stem cells (MSCs) have potent stabilising effects on vascular endothelium injury, inhibiting endothelial permeability in lung injury via paracrine hepatocyte growth factor (HGF). Recently, it has been indicated that MSCs secrete more factors by MSC-endothelial cell (MSC-EC) interactions. We hypothesised that MSC-EC interactions restore endothelial permeability induced by lipopolysaccharide (LPS) via paracrine HGF. Methods: We investigated the endothelial permeability induced by LPS under two co-culture conditions. Human pulmonary microvascular endothelial cells (HPMECs) were added into the upper chambers of cell-culture inserts, while two different co-culture conditions were used in the lower side of the transwells, as follows: (1) MSC-EC interaction group: MSCs and HPMECs contact co-culture; (2) MSC group: MSCs only. The endothelial paracellular and transcellular permeabilities in the upper side of transwells were detected. Then the concentration of HGF was measured in the culture medium by using an enzyme-linked immunosorbent assay kit, followed by neutralisation of HGF with anti-HGF antibody in the co-culture medium. In addition, adherens junction and cytoskeleton protein expressions were measured by Western blot and immunofluorescence. HPMEC proliferation was analysed by bromodeoxyuridine incorporation assay. Results: The paracellular permeability significantly increased after LPS stimulation in a dose-dependent and time-dependent manner. Meanwhile, MSC-EC interaction more significantly decreased endothelial paracellular and transcellular permeability induced by LPS. Moreover, HGF levels in the MSC-EC interaction group were much higher than those of the MSC group. However, neutralising HGF with anti-HGF antibody inhibited the role of MSC-EC interaction in improving endothelial permeability. Compared with the MSC group, MSC-EC interaction increased vascular endothelial (VE)-cadherin and occludin protein expression, reduced caveolin-1 protein expression in HPMECs, and restored remodelling of F-actin and junctional localisation of VE-cadherin. Furthermore, the proliferation ratio in the MSC-EC interaction group was higher than that of the MSC group. However, the effects of MSCs were significantly blocked by anti-HGF antibody. Conclusions: These data suggested that MSC-EC interaction decreased endothelial permeability induced by LPS, which was attributed mainly to HGF secreted by MSCs. The main mechanisms by which HGF restored the integrity of endothelial monolayers were remodelling of endothelial intercellular junctions, decreasing caveolin-1 protein expression, and inducing proliferation in HPMECs. Less
Abstract: The efficient delivery of chemotherapeutics to the tumor via nanoparticle (NP)-based delivery systems remains a significant challenge. This is compounded by the... More
Abstract: The efficient delivery of chemotherapeutics to the tumor via nanoparticle (NP)-based delivery systems remains a significant challenge. This is compounded by the fact that the tumor is highly dynamic and complex environment composed of a plurality of cell types and extracellular matrix. Since glycosaminoglycan (GAG) production is altered in many diseases (or pathologies), NPs bearing GAG moieties on the surface may confer some unique advantages in interrogating the tumor microenvironment. In order to explore this premise, in the study reported here poly-lactide-co-glycolide (PLGA) NPs in the range of 100–150 nm bearing various proteoglycans were synthesized by a single-step nanoprecipitation and characterized. The surface functionalization of the NPs with GAG moieties was verified using zeta potential measurements and X-ray photoelectron spectroscopy. To establish these GAG-bearing NPs as carriers of therapeutics, cellular toxicity assays were undertaken in lung epithelial adenocarcinoma (A549) cells, human pulmonary microvascular endothelial cells (HPMEC), and renal proximal tubular epithelial cells. In general NPs were well tolerated over a wide concentration range (100–600 µg/mL) by all cell types and were taken up to appreciable extents without any adverse cell response in A549 cells and HPMEC. Further, GAG-functionalized PLGA NPs were taken up to different extents in A459 cells and HPMEC. In both cell systems, the uptake of heparin-modified NPs was diminished by 50%–65% in comparison to that of unmodified PLGA. Interestingly, the uptake of chondroitin sulfate NPs was the highest in both cell systems with 40%–60% higher uptake when compared with that of PLGA, and this represented an almost twofold difference over heparin-modified NPs. These findings suggest that GAG modification can be explored as means of changing the uptake behavior of PLGA NPs and these NP systems have potential in cancer therapy. Keywords: tumor microenvironment, nanocarriers, proteoglycans, polysaccharides, passive targeting Less
Adhesion interactions between Plasmodium falciparum-infected erythrocytes (IE) and human cells underlie the pathology of severe malaria. IE cytoadhere to microvascular en... More
Adhesion interactions between Plasmodium falciparum-infected erythrocytes (IE) and human cells underlie the pathology of severe malaria. IE cytoadhere to microvascular endothelium or form rosettes with uninfected erythrocytes to survive in vivo by sequestering IE in the microvasculature and avoiding splenic clearance mechanisms. Both rosetting and cytoadherence are mediated by the parasite-derived IE surface protein family Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1). Rosetting and cytoadherence have been widely studied as separate entities; however, the ability of rosetting P. falciparum strains to cytoadhere has received little attention. Here, we show that IE of the IT/R29 strain expressing a rosette-mediating PfEMP1 variant (IT4var09) cytoadhere in vitro to a human brain microvascular endothelial cell line (HBEC-5i). Cytoadherence was inhibited by heparin and by treatment of HBEC-5i with heparinase III, suggesting that the endothelial receptors for IE binding are heparan sulfate proteoglycans. Antibodies to the N-terminal regions of the IT4var09 PfEMP1 variant (NTS-DBL1α and DBL2γ domains) specifically inhibited and reversed cytoadherence down to low concentrations (<10 μg/ml of total IgG). Surface plasmon resonance experiments showed that the NTS-DBLα and DBL2γ domains bind strongly to heparin, with half-maximal binding at a concentration of ∼0.5 μM in both cases. Therefore, cytoadherence of IT/R29 IE is distinct from rosetting, which is primarily mediated by NTS-DBL1α interactions with complement receptor 1. These data show that IT4var09-expressing parasites are capable of dual interactions with both endothelial cells and uninfected erythrocytes via distinct receptor-ligand interactions. Less
Adhesion interactions between Plasmodium falciparum-infected erythrocytes (IE) and human cells underlie the pathology of severe malaria. IE cytoadhere to microvascular en... More
Adhesion interactions between Plasmodium falciparum-infected erythrocytes (IE) and human cells underlie the pathology of severe malaria. IE cytoadhere to microvascular endothelium or form rosettes with uninfected erythrocytes to survive in vivo by sequestering IE in the microvasculature and avoiding splenic clearance mechanisms. Both rosetting and cytoadherence are mediated by the parasite-derived IE surface protein family Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1). Rosetting and cytoadherence have been widely studied as separate entities; however, the ability of rosetting P. falciparum strains to cytoadhere has received little attention. Here, we show that IE of the IT/R29 strain expressing a rosette-mediating PfEMP1 variant (IT4var09) cytoadhere in vitro to a human brain microvascular endothelial cell line (HBEC-5i). Cytoadherence was inhibited by heparin and by treatment of HBEC-5i with heparinase III, suggesting that the endothelial receptors for IE binding are heparan sulfate proteoglycans. Antibodies to the N-terminal regions of the IT4var09 PfEMP1 variant (NTS-DBL1α and DBL2γ domains) specifically inhibited and reversed cytoadherence down to low concentrations (<10 μg/ml of total IgG). Surface plasmon resonance experiments showed that the NTS-DBLα and DBL2γ domains bind strongly to heparin, with half-maximal binding at a concentration of ∼0.5 μM in both cases. Therefore, cytoadherence of IT/R29 IE is distinct from rosetting, which is primarily mediated by NTS-DBL1α interactions with complement receptor 1. These data show that IT4var09-expressing parasites are capable of dual interactions with both endothelial cells and uninfected erythrocytes via distinct receptor-ligand interactions. Less
Cerebral malaria is the most deadly manifestation of infection with Plasmodium falciparum. The pathology of cerebral malaria is characterized by the accumulation of infec... More
Cerebral malaria is the most deadly manifestation of infection with Plasmodium falciparum. The pathology of cerebral malaria is characterized by the accumulation of infected erythrocytes (IEs) in the microvasculature of the brain caused by parasite adhesins on the surface of IEs binding to human receptors on microvascular endothelial cells. The parasite and host molecules involved in this interaction are unknown. We selected three P. falciparum strains (HB3, 3D7, and IT/FCR3) for binding to a human brain endothelial cell line (HBEC-5i). The whole transcriptome of isogenic pairs of selected and unselected parasites was analyzed using a variant surface antigen-supplemented microarray chip. After selection, the most highly and consistently up-regulated genes were a subset of group A-like var genes (HB3var3, 3D7_PFD0020c, ITvar7, and ITvar19) that showed 11- to >100-fold increased transcription levels. These var genes encode P. falciparum erythrocyte membrane protein (PfEMP)1 variants with distinct N-terminal domain types (domain cassette 8 or domain cassette 13). Antibodies to HB3var3 and PFD0020c recognized the surface of live IEs and blocked binding to HBEC-5i, thereby confirming the adhesive function of these variants. The clinical in vivo relevance of the HBEC-selected parasites was supported by significantly higher surface recognition of HBEC-selected parasites compared with unselected parasites by antibodies from young African children suffering cerebral malaria (Mann-Whitney test, P = 0.029) but not by antibodies from controls with uncomplicated malaria (Mann-Whitney test, P = 0.58). This work describes a binding phenotype for virulence-associated group A P. falciparum erythrocyte membrane protein 1 variants and identifies targets for interventions to treat or prevent cerebral malaria. Less
Cardiovascular deconditioning is known to occur in astronauts exposed to microgravity. Endothelial dysfunction at microcirculatory sites might contribute to cardiovascula... More
Cardiovascular deconditioning is known to occur in astronauts exposed to microgravity. Endothelial dysfunction at microcirculatory sites might contribute to cardiovascular deconditioning induced by weightlessness. Recent studies have reported changes in the morphology and gene expression of endothelial cells exposed to conditions of simulated microgravity. The present study was aimed at examining the effects of microgravity on the apoptosis of microvascular endothelial cells and the mechanism underlying these effects. We simulated a microgravity environment and found that microgravity induced microvascular endothelial cell apoptosis and that this effect was correlated with the downregulation of the PI3K/Akt pathway, increased expression of NF-κB, and depolymerization of F-actin. These findings may provide important insights into the origin of the adverse physiological changes occurring due to exposure to microgravity conditions. Less
Phosphoinositide 3-kinase gamma(PI3Kgamma) is a critical mediator of directional cell movement. Here, we sought to characterise the role of PI3Kgamma in mediating the dif... More
Phosphoinositide 3-kinase gamma(PI3Kgamma) is a critical mediator of directional cell movement. Here, we sought to characterise the role of PI3Kgamma in mediating the different steps of polymorphonuclear leukocyte (PMN) trafficking in the lung. In a murine model of lipopolysaccharide (LPS)-induced lung injury, PMN migration into the different lung compartments was determined in PI3Kgamma gene-deficient (PI3Kgamma(-/-)) and wild-type mice. Bone marrow chimeras were created to characterise the role of PI3Kgamma on haematopoietic versus nonhaematopoietic cells. A small-molecule PI3Kgamma inhibitor was tested in vitro and in vivo. PMN adhesion to the pulmonary endothelium and transendothelial migration into the lung interstitium was enhanced in PI3Kgamma(-/-) mice. However, transepithelial migration into the alveolar space was reduced in these mice. When irradiated PI3Kgamma(-/-) mice were reconstituted with bone marrow from wild-type mice, migratory activity into the alveolar space was restored partially. A small-molecule PI3Kgamma inhibitor reduced chemokine-induced PMN migration in vitro when PMNs or epithelial cells, but not when endothelial cells, were treated. The inhibitor also reduced LPS-induced PMN migration in vivo. We conclude that PI3Kgamma is required for transepithelial but not for transendothelial migration in LPS-induced lung injury. Inhibition of PI3Kgamma activity may be effective at curbing excessive PMN infiltration in lung injury. Less
Although inflammation and altered barrier functions of the vasculature, due predominantly to the infection of endothelial cell lining of small and medium-sized blood vess... More
Although inflammation and altered barrier functions of the vasculature, due predominantly to the infection of endothelial cell lining of small and medium-sized blood vessels, represent salient pathological features of human rickettsioses, the interactions between pathogenic rickettsiae and microvascular endothelial cells remain poorly understood. We have investigated the activation of nuclear transcription factor-kappa B (NF-κB) and p38 mitogen-activated protein (MAP) kinase, expression of heme oxygenase 1 (HO-1) and cyclooxygenase 2 (COX-2), and secretion of chemokines and prostaglandins after Rickettsia rickettsii infection of human cerebral, dermal, and pulmonary microvascular endothelial cells in comparison with pulmonary artery cells of macrovascular origin. NF-κB and p38 kinase activation and increased HO-1 mRNA expression were clearly evident in all cell types, along with relatively similar susceptibility to R. rickettsii infection in vitro but considerable variations in the intensities/kinetics of the aforementioned host responses. As expected, the overall activation profiles of macrovascular endothelial cells derived from human pulmonary artery and umbilical vein were nearly identical. Interestingly, cerebral endothelial cells displayed a marked refractoriness in chemokine production and secretion, while all other cell types secreted various levels of interleukin-8 (IL-8) and monocyte chemoattractant protein 1 (MCP-1) in response to infection. A unique feature of all microvascular endothelial cells was the lack of induced COX-2 expression and resultant inability to secrete prostaglandin E2 after R. rickettsii infection. Comparative evaluation thus yields the first experimental evidence for the activation of both common and unique cell type-specific host response mechanisms in macrovascular and microvascular endothelial cells infected with R. rickettsii, a prototypical species known to cause Rocky Mountain spotted fever in humans. Less
Although the liver is known to be the main site of factor VIII (FVIII) production, other organs are probably also important for the regulation of FVIII secretion. However... More
Although the liver is known to be the main site of factor VIII (FVIII) production, other organs are probably also important for the regulation of FVIII secretion. However, the study of the regulation of extrahepatic FVIII production has been hampered by the lack of definitive identification of human tissues able to secrete FVIII. Recent studies have shown that lung endothelial cells can synthesize FVIII. We therefore studied the production of FVIII by endothelial cells purified from other vascular beds. Because physiologic stress results in a rapid elevation of FVIII, we also investigated whether endothelial cells can store FVIII and secrete it after treatment with agonists. Microvascular endothelial cells from lung, heart, intestine, and skin as well as endothelial cells from pulmonary artery constitutively secreted FVIII and released it after treatment with phorbol-myristate acetate and epinephrine. By contrast, endothelial cells from the aorta, umbilical artery and umbilical vein did not constitutively secrete FVIII or release it after treatment with agonists, probably because of a lack of FVIII synthesis. Extrahepatic endothelial cells from certain vascular beds therefore appear to be an important FVIII production and storage site with the potential to regulate FVIII secretion in chronic and acute conditions. Less
Inhaled nitric oxide may be protective against hyperoxic injury in the premature lung, but the mechanism is unknown. We hypothesized that nitric oxide (NO) would prevent ... More
Inhaled nitric oxide may be protective against hyperoxic injury in the premature lung, but the mechanism is unknown. We hypothesized that nitric oxide (NO) would prevent hyperoxia-induced NF-κB activation in neonatal pulmonary microvascular endothelial cells (HPMEC) and prevent the upregulation of target genes. Following hyperoxic exposure (O2> 95%), nuclear NF-κB consensus sequence binding increased and was associated with IκBα degradation. Both of these findings were prevented by exposure to NO. Furthermore, ICAM-1 mRNA and protein levels increased in cells exposed to hyperoxia, an effect abrogated by NO. To evaluate the potentially toxic effect of NO plus hyperoxia, cell viability and proliferation were assessed. Cells exposed to NO plus hyperoxia demonstrated improved survival as measured by trypan blue exclusion when compared to cells exposed to hyperoxia alone. These differences in cell death could not be attributed to apoptosis measured by caspase-3 activity. Finally, cellular proliferation inhibited by hyperoxia was rescued by concurrent exposure to NO. These data demonstrate that NO prevents hyperoxia-induced NF-κB activation in HPMEC and results in decreased expression of adhesion molecules and decreased cellular toxicity. This may help explain the protective effects of NO on hyperoxic injury in the developing lung vasculature. Less
IL-6 is a biological marker of ventilator-associated lung injury that may contribute to alveolar barrier dysfunction in acute respiratory distress syndrome. To determine ... More
IL-6 is a biological marker of ventilator-associated lung injury that may contribute to alveolar barrier dysfunction in acute respiratory distress syndrome. To determine whether IL-6 affects alveolar barrier disruption in a model of ventilator-induced lung injury, we examined alveolar barrier albumin flux in wild-type (WT) mice given an IL-6-blocking Ab (IL6AB) and mice deficient in IL-6 (IL6KO). Albumin flux was significantly higher in mice given IL6AB compared with mice given a control Ab. Unexpectedly, albumin flux was similar in WT and IL6KO mice. To examine the mechanisms for these findings, lung neutrophil accumulation (myeloperoxidase activity) was compared, revealing a correlation between lung neutrophil accumulation and albumin flux. IL6AB mice had significantly more lung neutrophils than WT and IL6KO mice, which were similar. Therefore, to determine whether the cellular source of IL-6 influences neutrophil accumulation and alveolar barrier function, chimeric mice were compared. WT/KO chimeras (WT mice with IL6KO hematopoietic cells) showed significantly greater albumin flux and neutrophil accumulation with mechanical ventilation than WT/WT mice. Neutrophil depletion decreased albumin flux in WT and WT/KO mice. IL6KO neutrophils were more adherent in an in vitro assay compared with WT neutrophils. IL-6 from a hematopoietic cell source limits alveolar barrier disruption potentially by reducing neutrophil contact with the endothelium. Modulation of IL-6 signaling in a cell type-specific fashion may be a therapeutic target for patients with acute lung injury. Less
Although current H5N1 highly pathogenic avian influenza viruses (HPAIV) are inefficiently transmitted to humans, infected individuals can suffer from severe disease, ofte... More
Although current H5N1 highly pathogenic avian influenza viruses (HPAIV) are inefficiently transmitted to humans, infected individuals can suffer from severe disease, often progressing rapidly to acute respiratory distress syndrome and multiorgan failure. This is in contrast with the situation with human influenza viruses, which in immunocompetent individuals usually cause only a respiratory disease which is less aggressive than that observed with avian H5N1 viruses. While the biological basis of inefficient transmission is well documented, the mechanisms by which the H5N1 viruses cause fatal disease remain unclear. In the present study, we demonstrate that human pulmonary microvascular endothelial cells (hPMEC) had a clearly higher susceptibility to infection by H5N1 HPAIV than to infection by human influenza viruses. This was measurable by de novo intracellular nucleoprotein production and virus replication. It was also related to a relatively higher binding capacity to cellular receptors. After infection of hPMEC, cell activation markers E-selectin and P-selectin were upregulated, and the proinflammatory cytokines interleukin-6 and beta interferon were secreted. H5N1 virus infection was also associated with an elevated rate of cell death. Reverse genetics analyses demonstrated a major role for the viral hemagglutinin in this cell tropism. Overall, avian H5N1 viruses have a particular receptor specificity targeting endothelial cells that is different from human influenza viruses, and this H5N1 receptor specificity could contribute to disease pathogenesis. Less
Microvascular endothelial cell (MVEC) injury coupled to progression of platelet microthrombi facilitated by ADAMTS13 deficiency is characteristic of idiopathic and HIV-li... More
Microvascular endothelial cell (MVEC) injury coupled to progression of platelet microthrombi facilitated by ADAMTS13 deficiency is characteristic of idiopathic and HIV-linked thrombotic thrombocytopenic purpura (TTP). Cytokines capable of inducing MVEC apoptosis in vitro are up-regulated in both TTP and HIV infection. However, the concentrations of these cytokines required to elicit EC apoptosis in vitro are 2- to 3-log-fold greater than present in patient plasmas. We report that clinically relevant levels of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and interferon (IFN)-gamma act in synergy to induce apoptosis in dermal MVECs, but have no effect on large-vessel ECs or pulmonary MVECs. This reflects the tissue distribution of TTP lesions in vivo. Sensitivity to TTP plasma or TRAIL plus IFN-gamma is paralleled by enhanced ubiquitination of the caspase-8 regulator cellular FLICE-like inhibitory protein (c-FLIP), targeting it for proteasome degradation. c-FLIP silencing with anti-FLIP short interfering RNA (siRNA) in pulmonary MVECs rendered them susceptible to TTP plasma- and cytokine-mediated apoptosis, while up-regulation of c-FLIP by gene transfer partially protected dermal MVECs from such injury. TTP plasma-mediated apoptosis appears to involve cytokine-induced acceleration of c-FLIP degradation, sensitizing cells to TRAIL-mediated caspase-8 activation and cell death. Suppression of TRAIL or modulation of immunoproteasome activity may have therapeutic relevance in TTP. Less
Changes in endothelial permeability are crucial in the pathogenesis of many diseases. Adenosine is one of the endogenous mediators controlling endothelial permeability un... More
Changes in endothelial permeability are crucial in the pathogenesis of many diseases. Adenosine is one of the endogenous mediators controlling endothelial permeability under normal conditions, and an endothelial cell surface enzyme CD73 is a key regulator of adenosine production. Here we report that IFN-beta is a novel inducer of CD73. We found that pretreatment with IFN-beta dramatically improved the vascular barrier function in lungs after intestinal ischemia-reperfusion injury in wild-type animals in vivo. IFN-beta had absolutely no protective effects in CD73-deficient mice, which suffered from more severe lung damage than wild-type mice, showing that IFN-beta functions strictly in a CD73-dependent manner. Most importantly, IFN-beta treatment initiated after the ischemic period almost completely inhibited vascular leakage during the reperfusion. IFN-beta also induced the expression and activity of CD73 and concurrently decreased vascular permeability in cultured human pulmonary endothelial cells. These data show that induction of CD73 and improvement of vascular barrier are new mechanisms for the anti-inflammatory action of IFN-beta. Moreover, IFN-beta treatment may be useful in alleviating vascular leakage induced by ischemia-reperfusion injury. Less
Increases in endothelial cGMP prevent oxidant-mediated endothelial barrier dysfunction, but the downstream mechanisms remain unclear. To determine the role of cGMP-depend... More
Increases in endothelial cGMP prevent oxidant-mediated endothelial barrier dysfunction, but the downstream mechanisms remain unclear. To determine the role of cGMP-dependent protein kinase (PKG)(I), human pulmonary artery endothelial cells (HPAEC) lacking PKG(I) expression were infected with a recombinant adenovirus encoding PKG(Ibeta) (Ad.PKG) and compared with uninfected and control-infected (Ad.betagal) HPAEC. Transendothelial electrical resistance (TER), an index of permeability, was measured after H(2)O(2) (250 microM) exposure with or without pretreatment with 8-(4-chlorophenylthio)guanosine 3',5'-cyclic monophosphate (CPT-cGMP). HPAEC infected with Ad.PKG, but not Ad.betagal, expressed PKG(I) protein and demonstrated Ser(239) and Ser(157) phosphorylation of vasodilator-stimulated phosphoprotein after treatment with CPT-cGMP. Adenoviral infection decreased basal permeability equally in Ad.PKG- and Ad.betagal-infected HPAEC compared with uninfected cells. Treatment with CPT-cGMP (100 microM) caused a PKG(I)-independent decrease in permeability (8.2 +/- 0.6%). In all three groups, H(2)O(2) (250 microM) caused a similar approximately 35% increase in permeability associated with increased actin stress fiber formation, intercellular gaps, loss of membrane VE-cadherin, and increased intracellular Ca(2+) concentration ([Ca(2+)](i)). In uninfected and Ad.betagal-infected HPAEC, pretreatment with CPT-cGMP (100 microM) partially blocked the increased permeability induced by H(2)O(2). In Ad.PKG-infected HPAEC, CPT-cGMP (50 microM) prevented the H(2)O(2)-induced TER decrease, cytoskeletal rearrangement, and loss of junctional VE-cadherin. CPT-cGMP attenuated the peak [Ca(2+)](i) caused by H(2)O(2) similarly (23%) in Ad.betagal- and Ad.PKG-infected HPAEC, indicating a PKG(I)-independent effect. These data suggest that cGMP decreased HPAEC basal permeability by a PKG(I)-independent process, whereas the ability of cGMP to prevent H(2)O(2)-induced barrier dysfunction was predominantly mediated by PKG(I) through a Ca(2+)-independent mechanism. Less
Pulmonary hypertension associated with human immunodeficiency virus (HIV) infection also involves injury to the lung endothelium. However, the pathogenesis of HIV-induced... More
Pulmonary hypertension associated with human immunodeficiency virus (HIV) infection also involves injury to the lung endothelium. However, the pathogenesis of HIV-induced pulmonary hypertension is not known; we hypothesized that HIV or secreted viral proteins could play a role in vascular injury and the increased frequency of pulmonary hypertension observed in HIV-infected patients. Here, we report that exposure of HIV-1 gp120 proteins to primary human lung microvascular endothelial cells causes apoptosis, as assessed by TUNEL assay, Annexin-V staining, and DNA laddering. Using ribonuclease protection assay and Western blotting we find that gp120-induced apoptosis of lung endothelial cells involves a down-regulation in Bcl-xl mRNA and proteins. In addition, gp120 significantly increases secretion of the potent vasoconstrictor endothelin-1 by human lung endothelial cells. These data suggest that secreted HIV gp120 proteins induce lung endothelial cell injury and could contribute to the development of HIV-associated pulmonary hypertension. Less