Rationale
Alveolar epithelial cell loss is a hallmark of idiopathic pulmonary fibrosis (IPF). Type I alveolar epithelial cells (AT1) line the lung alveoli to facilitate ... More
Rationale
Alveolar epithelial cell loss is a hallmark of idiopathic pulmonary fibrosis (IPF). Type I alveolar epithelial cells (AT1) line the lung alveoli to facilitate gas exchange. In IPF, repeat injury leads to AT1 cell loss, releasing pro-inflammatory factors. In preclinical models, including bleomycin, AT1 cells are lost through apoptosis, an effect that is diminished in LPA1-/- mice. Here, we wanted to determine whether, 1) LPA directly mediated human AT1 apoptosis through LPA1 and 2) conditioned media from human IPF-derived precision cut lung slice (PCLS) cultures could induce apoptosis in an LPA1-sensitive manner.
Methods
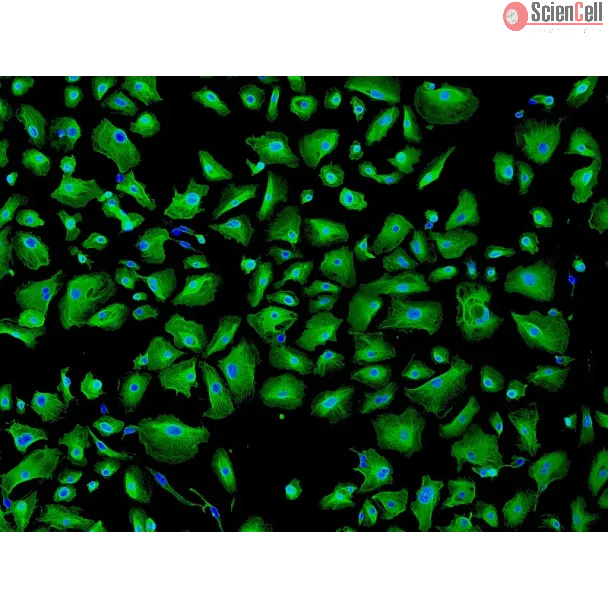
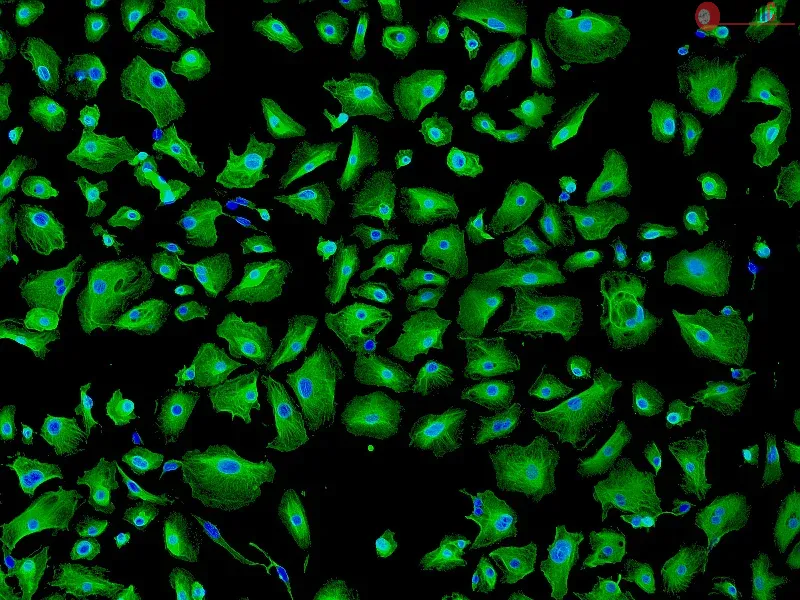
Human primary epithelial cells (HPAEpiCs, ScienCell) were plated at 15k/96-well and adhered overnight. In culture, HPAEpiCs terminally differentiate into AT1 cells. Cells were pretreated with vehicle or 300 nM PIPE-791 for 8h. LPA was added to the cells, incubated for 24h, then immunostained for podoplanin (Pdpn), cleaved caspase 3 (Asp175; Cc3), phalloidin, and Hoechst. Apoptosis was measured as a %Cc3+/Pdpn+/Hoechst+ of total Pdpn+/Hoechst+ counts. For conditioned media studies, PCLS were cultured in vehicle or 300 nM PIPE-791. Conditioned media was collected at study end and frozen (-80oC). Separately, AT1 cells were preincubated with vehicle or PIPE-791 for 8h, followed by 50% exchange of culture media for conditioned media. If needed, PIPE-791 was added to reach a 300 nM final concentration.
Results
Direct addition of LPA significantly increased the %Cc3+/Pdpn+ cells, indicating apoptosis. This increase was inhibited by PIPE-791. AT1 cells exposed to normal-PCLS conditioned media did not show a significant difference in the %Cc3+/Pdpn+ cells compared to complete growth media. Exposure of AT1 cells to IPF-PCLS conditioned media, however, significantly increased Cc3+/Pdpn+ cells and was inhibited by PIPE-791. Interestingly, conditioned media from PIPE-791 treated IPF-PCLS cultures did not increase the %Cc3+/Pdpn+. AT1 differentiation was not affected by any of the aforementioned treatments.
Conclusions
Alveolar epithelial cell loss is an established hallmark of IPF and may occur by way of apoptosis. Using cultured human AT1 cells, we determined that cleaved caspase 3 is induced by direct addition of either LPA or IPF-PCLS conditioned media, and prevented by the LPA1-selective antagonist, PIPE-791. Of note, conditioned media from IPF-PCLS cultures treated with PIPE-791 did not increase the %Cc3+/Pdpn+ cells, suggesting LPA1 is involved in yet undefined pro-apoptotic factors released by the IPF-PCLS. Thus, while LPA1’s role in fibroblast activation is well-established, we postulate that antagonizing LPA1 prevents additional mechanisms involved in IPF initiation and progression. Less
Breast cancer and lung adenocarcinoma share common features, including female predominance and the expression of estrogen receptor (ER) and epidermal growth factor recept... More
Breast cancer and lung adenocarcinoma share common features, including female predominance and the expression of estrogen receptor (ER) and epidermal growth factor receptor (EGFR) during carcinogenesis. Patients with breast cancer have a significantly higher risk of developing second primary lung cancer than those without breast cancer. ER beta expression is associated with resistance to EGFR tyrosine kinase inhibitors (TKIs) in EGFR-mutant lung adenocarcinoma, indicating a potentially important interaction between ER and EGFR. However, the mechanisms underlying this crosstalk remain poorly understood. Our clinical data showed a significant correlation between antiestrogen treatment for breast cancer and mutant EGFR expression (p = 0.021) in lung adenocarcinoma patients. In vitro, tamoxifen upregulated phosphorylated EGFR (p-EGFR) in EGFR-mutant lung adenocarcinoma cell lines. Heparin-binding EGF-like growth factor was identified as a key mediator from the ER pathway that stimulates p-EGFR. Tamoxifen counteracts estrogen’s effect and restores p-EGFR upregulation. Furthermore, coadministration of tamoxifen and the EGFR TKI gefitinib potentially inhibited p-EGFR expression in EGFR-mutant lung adenocarcinoma. Regular follow-up with chest computed tomography is recommended for patients with breast cancer. For those diagnosed with both ER-positive breast cancer and EGFR-mutant lung adenocarcinoma, combined tamoxifen and EGFR TKI therapy may offer an effective targeted treatment strategy.
Keywords:
lung adenocarcinoma; epidermal growth factor receptor; estrogen receptor; breast cancer Less
Alveolar regeneration requires coordinated alveolar type 1 (AT1)/alveolar type 2 (AT2) responses and an intact epithelial barrier. In vitro systems, however, rarely coupl... More
Alveolar regeneration requires coordinated alveolar type 1 (AT1)/alveolar type 2 (AT2) responses and an intact epithelial barrier. In vitro systems, however, rarely couple lineage control with high resistance. Here we establish a concise workflow in which human AT2-like progenitors are expanded in a medium containing a small-molecule cocktail (Y-27632/A-83-01/CHIR99021; YAC), followed by air–liquid interface (ALI) culture in medium without the YAC cocktail (hereafter referred to as the YAC(−) condition). Compared with submerged culture, ALI increased AT1 (AQP5, AGER, and PDPN) and AT2 (SFTPB, SFTPC, and SFTPD) markers. Within ALI, β-catenin inhibition by XAV939 elevated AT1 features while retaining AT2 protein expression, but was accompanied by reduced AT2 marker expression at the transcriptional level compared with the YAC(−) condition. Transepithelial electrical resistance entered the kΩ range by ALI day 10 and remained high across ALI conditions, with hematoxylin and eosin staining confirming a continuous single-layer epithelium. Passage reduced absolute marker levels and resistance but preserved the rank order across conditions. In scratch assays, ALI monolayers closed wounds over 24–48 h, and spatial immunofluorescence staining showed surfactant protein-C enrichment at the leading edge and podoplanin predominantly behind the front. Collectively, these findings demonstrate that β-catenin modulation within ALI biases the alveolar epithelial profile toward AT1-like features, whereas the YAC(−) condition sustains a balanced AT1/AT2 state with superior epithelial barrier performance and repair capacity. This platform provides a single-monolayer system suitable for dissecting alveolar epithelial differentiation and functional maintenance.
Keywords: Air–liquid interface culture; β-catenin inhibition; Wnt signaling modulation; Transepithelial electrical resistance; Alveolar lineage plasticity; Epithelial repair
Less
Idiopathic pulmonary fibrosis (IPF) is a progressive disease with high mortality and limited therapeutic options. The immune checkpoint PD1/PD-L1 axis is related to the p... More
Idiopathic pulmonary fibrosis (IPF) is a progressive disease with high mortality and limited therapeutic options. The immune checkpoint PD1/PD-L1 axis is related to the pathogenesis of pulmonary fibrosis, and upregulated expression levels of PD-L1 have been demonstrated in IPF patients. However, the mechanism of PD-L1 in pulmonary fibrosis is not fully understood. Here, we demonstrated upregulated expression of PD-L1 in fibrotic lung tissues and sera of IPF patients. Bleomycin (BLM) treatment induced PD-L1 upregulation, EMT (Epithelial-Mesenchymal Transition) and fibrosis-like morphology changes in human pulmonary alveolar epithelial cells (HPAEpiCs). Silencing PD-L1 attenuated BLM-induced EMT and fibrosis-like morphology changes in HPAEpiCs. In addition, we identified that PD-L1 directly binds to vimentin and inhibits vimentin ubiquitination, thereby increasing vimentin levels in HPAEpiCs. Silencing of vimentin inhibited BLM- and PD-L1-induced fibrosis in HPAEpiCs. The correlation between PD-L1 and EMT or vimentin expression was further confirmed in clinical samples and animal models. Finally, we used BLM- and paraquat-induced pulmonary fibrosis animal models to confirm the anti-pulmonary fibrosis effects of PD-L1 silencing. Taken together, our findings suggest that upregulated PD-L1 stimulates EMT of alveolar epithelial cells by increasing vimentin levels by inhibiting vimentin ubiquitination, thereby contributing to pulmonary fibrosis. Less
Pulmonary fibrosis represents the terminal stage of a diverse group of lung diseases including scleroderma associated interstitial lung disease. The molecular mechanisms ... More
Pulmonary fibrosis represents the terminal stage of a diverse group of lung diseases including scleroderma associated interstitial lung disease. The molecular mechanisms underlying the pathogenesis of lung fibrosis are not well understood and there is a great need for more effective treatment for this lethal disease. We recently discovered a small fragment of hepatocyte growth factor (HGF) receptor MET as a peptide designated “M10,” with strong antifibrotic properties. Furthermore, we showed that aspartic acid at position 1398 of MET is essential for M10 generation. The current study was undertaken to investigate the D1398G variant of MET in which aspartic acid at position 1398 was mutated to glycine resulting in loss of M10. We demonstrate that lung fibroblasts, A549, and primary alveolar epithelial cells (AEC) expressing D1398G MET exhibit reduced auto-phosphorylation on tyrosine residues and reduced activation of Ras and MAPK. HGF treatment of scleroderma lung fibroblasts as well as HGF treatment of TGFβ-treated normal lung fibroblasts transfected with wild type MET is associated with decreased collagen, connective tissue growth factor (CTGF, CCN2) and smooth muscle α-actin (SMA). However, HGF has no such effects in cells transfected with MET D1398G. Cisplatin- and FasL-induced apoptosis is significantly reduced in AEC transfected with MET wild type, but not in AEC transfected with MET D1398G. We conclude that the D1398G variant of MET is associated with compromised phosphorylation and impaired HGF signaling in lung fibroblasts and AEC, two cell types implicated in the pathogenesis of pulmonary fibrosis associated with scleroderma. Ongoing studies will explore the frequency of this variant and its relationship to pulmonary outcomes in scleroderma patients. Less
The proinflammatory interleukin-33 (IL-33) binds to its receptor ST2L on the surface of immune cells and stimulates the production of Th2 cytokines; however, the effects ... More
The proinflammatory interleukin-33 (IL-33) binds to its receptor ST2L on the surface of immune cells and stimulates the production of Th2 cytokines; however, the effects of IL-33 on tumour cells are poorly understood. Here we show that ST2 was significantly downregulated in human lung cancer tissues and cells compared with normal lung tissues and cells. IL-33 expression was also inversely correlated with the stages of human lung cancers. In accordance with this finding, low-metastatic cells but not high-metastatic cells derived from Lewis lung carcinoma expressed functional ST2L. IL-33 was abundantly present in the tumours established by the low-metastatic cells compared with those formed by the high-metastatic cells. Although the low-metastatic cells scarcely expressed IL-33 in vitro, these cells did expry 6ess this molecule in vivo, likely due to stimulation by intratumoural IL-1β and IL-33. Importantly, IL-33 enhanced the cell death of ST2L-positive low-metastatic cells, but not of ST2L-negative high-metastatic cells, under glucose-depleted, glutamine-depleted and hypoxic conditions through p38 MAPK and mTOR activation, and in a mitochondria-dependent manner. The cell death was characterised by cytoplasmic blisters and karyolysis, which are unique morphological features of oncosis. Inevitably, the low-metastatic cells, but not of the high-metastatic cells, grew faster in IL-33−/− mice than in wild-type mice. Furthermore, IL-33 selected for the ST2L-positive, oncosis-resistant high-metastatic cells under conditions mimicking the tumour microenvironment. These data suggest that IL-33 enhances lung cancer progression by selecting for more malignant cells in the tumour microenvironment. Less
Postinfluenza pneumococcal pneumonia is a common cause of death in humans. However, the role of IL-27 in the pathogenesis of secondary pneumococcal pneumonia after influe... More
Postinfluenza pneumococcal pneumonia is a common cause of death in humans. However, the role of IL-27 in the pathogenesis of secondary pneumococcal pneumonia after influenza is unknown. We now report that influenza infection induced pulmonary IL-27 production in a type I IFN-α/β receptor (IFNAR) signalling-dependent manner, which sensitized mice to secondary pneumococcal infection downstream of IFNAR pathway. Mice deficient in IL-27 receptor were resistant to secondary pneumococcal infection and generated more IL-17A-producing γδ T cells but not αβ T cells, thereby leading to enhanced neutrophil response during the early phase of host defence. IL-27 treatment could suppress the development of IL-17A-producing γδ T cells activated by Streptococcus pneumoniae and dendritic cells. This suppressive activity of IL-27 on γδ T cells was dependent on transcription factor STAT1. Finally, neutralization of IL-27 or administration of IL-17A restored the role of γδ T cells in combating secondary pneumococcal infection. Our study defines what we believe to be a novel role of IL-27 in impairing host innate immunity against pneumococcal infection. Less
Postinfluenza pneumococcal pneumonia is a common cause of death in humans. However, the role of IL-27 in the pathogenesis of secondary pneumococcal pneumonia after influe... More
Postinfluenza pneumococcal pneumonia is a common cause of death in humans. However, the role of IL-27 in the pathogenesis of secondary pneumococcal pneumonia after influenza is unknown. We now report that influenza infection induced pulmonary IL-27 production in a type I IFN-α/β receptor (IFNAR) signalling-dependent manner, which sensitized mice to secondary pneumococcal infection downstream of IFNAR pathway. Mice deficient in IL-27 receptor were resistant to secondary pneumococcal infection and generated more IL-17A-producing γδ T cells but not αβ T cells, thereby leading to enhanced neutrophil response during the early phase of host defence. IL-27 treatment could suppress the development of IL-17A-producing γδ T cells activated by Streptococcus pneumoniae and dendritic cells. This suppressive activity of IL-27 on γδ T cells was dependent on transcription factor STAT1. Finally, neutralization of IL-27 or administration of IL-17A restored the role of γδ T cells in combating secondary pneumococcal infection. Our study defines what we believe to be a novel role of IL-27 in impairing host innate immunity against pneumococcal infection. Less
Xiao-Qing-Long-Tang (XQLT) is known to regulate allergic immune reactions. The aim of this study was to investigate the effects of XQLT on allergen-induced cytokines and ... More
Xiao-Qing-Long-Tang (XQLT) is known to regulate allergic immune reactions. The aim of this study was to investigate the effects of XQLT on allergen-induced cytokines and associated signaling pathways. An acute allergic mouse model was used to investigate the effects of XQLT on nerve growth factor (NGF) during an allergic reaction, while human pulmonary alveolar epithelial cells (HPAEpiCs) were used to investigate the effects of XQLT on Dermatophagoides pteronyssinus group 2 (Der p 2)-induced NGF, p75 neurotrophin receptor (p75NTR) and thymic stromal lymphopoietin (TSLP) expression. XQLT was demonstrated to inhibit NGF- and p75NTR-related allergic reactions in the mouse model. XQLT also reduced the expression of Toll-like receptor 4 (TLR4) in the lungs of the model mice. XQLT inhibited Der p 2-induced NGF, TSLP and p75NTR expression in the HPAEpiC cell line. The use of recombinant soluble TLR4 (sTLR4) in a competitive assay partially attenuated the inhibitory effect of XQLT on NGF, TSLP and p75NTR expression in HPAEpiC cells. The inhibitory effect of XQLT on the Ser536 phosphorylation of p65 (nuclear factor-κB; NF-κB), a TLR4-induced factor, was also attenuated by sTLR4. In conclusion, XQLT inhibited Der p allergen-induced NGF, p75NTR and TSLP expression. This inhibition was attenuated by sTLR4. The mechanism of action of XQLT may be correlated with TLR4, a primary receptor in the innate immune system. The findings of this study may focus the search for pharmacological targets of XQLT onto TLR4, which is important in the allergen presentation pathway. Less
Background: Protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme capable of methylating histone and non-histone proteins, and is involved in a wid... More
Background: Protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme capable of methylating histone and non-histone proteins, and is involved in a wide range of cellular processes that range from transcriptional regulation to organelle biosynthesis. As such, its overexpression has been linked to tumor suppressor gene silencing, enhanced tumor cell growth and survival. Material and methods: Quantitative real-time polymerase chain reaction, Western immunoblot and immunohistochemistry were used to characterize PRMT5 expression in lung cancer cell lines and human tumors. Clinicopathological findings of tissue microarray based samples from 229 patients with non-small cell lung carcinomas (NSCLC) and 133 cases with pulmonary neuroendocrine tumors (NET) were analyzed with regard to nuclear and cytoplasmic PRMT5 expression. Results: There was statistically significant difference in PRMT5 messenger RNA expression between tumors and nonneoplastic lung tissues. Immunoblot experiments showed abundant expression of PRMT5 and its symmetric methylation mark H4R3 in lung carcinoma but not in non-neoplastic human pulmonary alveolar and bronchial epithelial cell lines. More than two thirds of lung tumors expressed PRMT5. High levels of cytoplasmic PRMT5 were detected in 20.5% of NSCLC and in 16.5% of NET; high levels of nuclear PRMT5 were detected in 38.0% of NSCLC and 24.0% of NET. Cytoplasmic PRMT5 was associated with high grade in both NSCLC and pulmonary NET while nuclear PRMT5 was more frequent in carcinoid tumors (p < 0.05). Conclusion: The observed findings support the role of PRMT5 in lung tumorigenesis and reflect its functional dichotomy in cellular compartments. Virtual slide: The virtual slides for this article can be found here: http://www.diagnosticpathology.diagnomx.eu/vs/1611895162102528. Less
Xiao-Qing-Long-Tang (XQLT) is known to regulate allergic immune reactions. The aim of this study was to investigate the effects of XQLT on allergen-induced cytokines and ... More
Xiao-Qing-Long-Tang (XQLT) is known to regulate allergic immune reactions. The aim of this study was to investigate the effects of XQLT on allergen-induced cytokines and associated signaling pathways. An acute allergic mouse model was used to investigate the effects of XQLT on nerve growth factor (NGF) during an allergic reaction, while human pulmonary alveolar epithelial cells (HPAEpiCs) were used to investigate the effects of XQLT on Dermatophagoides pteronyssinus group 2 (Der p 2)-induced NGF, p75 neurotrophin receptor (p75NTR) and thymic stromal lymphopoietin (TSLP) expression. XQLT was demonstrated to inhibit NGF- and p75NTR-related allergic reactions in the mouse model. XQLT also reduced the expression of Toll-like receptor 4 (TLR4) in the lungs of the model mice. XQLT inhibited Der p 2-induced NGF, TSLP and p75NTR expression in the HPAEpiC cell line. The use of recombinant soluble TLR4 (sTLR4) in a competitive assay partially attenuated the inhibitory effect of XQLT on NGF, TSLP and p75NTR expression in HPAEpiC cells. The inhibitory effect of XQLT on the Ser536 phosphorylation of p65 (nuclear factor-κB; NF-κB), a TLR4-induced factor, was also attenuated by sTLR4. In conclusion, XQLT inhibited Der p allergen-induced NGF, p75NTR and TSLP expression. This inhibition was attenuated by sTLR4. The mechanism of action of XQLT may be correlated with TLR4, a primary receptor in the innate immune system. The findings of this study may focus the search for pharmacological targets of XQLT onto TLR4, which is important in the allergen presentation pathway. Less
Cytosolic phospholipase A2 (cPLA2) plays a pivotal role in mediating agonist-induced arachidonic acid (AA) release for prostaglandin (PG) synthesis during inflammation tr... More
Cytosolic phospholipase A2 (cPLA2) plays a pivotal role in mediating agonist-induced arachidonic acid (AA) release for prostaglandin (PG) synthesis during inflammation triggered by tumor necrosis factor-α (TNF-α). However, the mechanisms underlying TNF-α-induced cPLA2 expression in human lung epithelial cells (HPAEpiCs) were not completely understood. Less
Background: Protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme capable of methylating histone and non-histone proteins, and is involved in a wid... More
Background: Protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme capable of methylating histone and non-histone proteins, and is involved in a wide range of cellular processes that range from transcriptional regulation to organelle biosynthesis. As such, its overexpression has been linked to tumor suppressor gene silencing, enhanced tumor cell growth and survival. Material and methods: Quantitative real-time polymerase chain reaction, Western immunoblot and immunohistochemistry were used to characterize PRMT5 expression in lung cancer cell lines and human tumors. Clinicopathological findings of tissue microarray based samples from 229 patients with non-small cell lung carcinomas (NSCLC) and 133 cases with pulmonary neuroendocrine tumors (NET) were analyzed with regard to nuclear and cytoplasmic PRMT5 expression. Results: There was statistically significant difference in PRMT5 messenger RNA expression between tumors and nonneoplastic lung tissues. Immunoblot experiments showed abundant expression of PRMT5 and its symmetric methylation mark H4R3 in lung carcinoma but not in non-neoplastic human pulmonary alveolar and bronchial epithelial cell lines. More than two thirds of lung tumors expressed PRMT5. High levels of cytoplasmic PRMT5 were detected in 20.5% of NSCLC and in 16.5% of NET; high levels of nuclear PRMT5 were detected in 38.0% of NSCLC and 24.0% of NET. Cytoplasmic PRMT5 was associated with high grade in both NSCLC and pulmonary NET while nuclear PRMT5 was more frequent in carcinoid tumors (p < 0.05). Conclusion: The observed findings support the role of PRMT5 in lung tumorigenesis and reflect its functional dichotomy in cellular compartments. Virtual slide: The virtual slides for this article can be found here: http://www.diagnosticpathology.diagnomx.eu/vs/1611895162102528. Less
While acute lung injury (ALI) contributes significantly to critical illness, it resolves spontaneously in many instances. The majority of patients experiencing ALI requir... More
While acute lung injury (ALI) contributes significantly to critical illness, it resolves spontaneously in many instances. The majority of patients experiencing ALI require mechanical ventilation. Therefore, we hypothesized that mechanical ventilation and concomitant stretch-exposure of pulmonary epithelia could activate endogenous pathways important in lung protection. Methods and Findings: To examine transcriptional responses during ALI, we exposed pulmonary epithelia to cyclic mechanical stretch conditions—an in vitro model resembling mechanical ventilation. A genome-wide screen revealed a transcriptional response similar to hypoxia signaling. Surprisingly, we found that stabilization of hypoxia-inducible factor 1A (HIF1A) during stretch conditions in vitro or during ventilator-induced ALI in vivo occurs under normoxic conditions. Extension of these findings identified a functional role for stretch-induced inhibition of succinate dehydrogenase (SDH) in mediating normoxic HIF1A stabilization, concomitant increases in glycolytic capacity, and improved tricarboxylic acid (TCA) cycle function. Pharmacologic studies with HIF activator or inhibitor treatment implicated HIF1A-stabilization in attenuating pulmonary edema and lung inflammation during ALI in vivo. Systematic deletion of HIF1A in the lungs, endothelia, myeloid cells, or pulmonary epithelia linked these findings to alveolar-epithelial HIF1A. In vivo analysis of 13C-glucose metabolites utilizing liquid-chromatography tandem mass-spectrometry demonstrated that increases in glycolytic capacity, improvement of mitochondrial respiration, and concomitant attenuation of lung inflammation during ALI were specific for alveolar-epithelial expressed HIF1A. Conclusions: These studies reveal a surprising role for HIF1A in lung protection during ALI, where normoxic HIF1A stabilization and HIF-dependent control of alveolar-epithelial glucose metabolism function as an endogenous feedback loop to dampen lung inflammation. Less
The etiology of chronic obstructive pulmonary disease (COPD) remains unclear. A mechanism involving the autoimmune reaction in the pathogenesis of COPD has been proposed ... More
The etiology of chronic obstructive pulmonary disease (COPD) remains unclear. A mechanism involving the autoimmune reaction in the pathogenesis of COPD has been proposed but not confirmed. The aim of this study was to investigate whether serum autoantibodies against pulmonary cellular proteins are present in COPD patients and to identify their autoantigens if possible. Samples from 50 COPD patients and 42 control subjects were studied. Circulating autoantibodies were detected by Western blot. Immunoprecipitation and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry were used to identify the autoantigens. Autoantibodies against pulmonary cellular antigens were found in the sera of COPD patients. Specifically, an autoantibody against the 45-kDa human cytokeratin 18 protein was found in 76.0% of COPD patients and 23.8% of control subjects (p<0.001). Furthermore, the cytokeratin 18 autoantibody level was positively correlated with the FEV(1) (L) (p=0.013) and FEV(1) (%pred.) (p=0.043) values observed in COPD patients. This study identified the pulmonary epithelial cytokeratin 18 protein as a COPD-associated autoantigen and found that anti-cytokeratin 18 autoantibodies were prevalent in COPD patients. Our results support the hypothesis that humoral autoimmunity may be involved in the pathogenesis of COPD. Copyright (c) 2009 Elsevier B.V. All rights reserved. Less
Epstein-Barr virus (EBV) is invariably present in undifferentiated nasopharyngeal carcinomas, is found sporadically in other carcinomas, and replicates in the differentia... More
Epstein-Barr virus (EBV) is invariably present in undifferentiated nasopharyngeal carcinomas, is found sporadically in other carcinomas, and replicates in the differentiated layer of the tongue epithelium in lesions of oral hairy leukoplakia. However, it is not clear how frequently or by what mechanism EBV infects epithelial cells normally. Here, we report that a human epithelial cell line, 293, can be stably infected by EBV that has been genetically marked with a selectable gene. We show that 293 cells express a relatively low level of CD21, that binding of fluorescein-labeled EBV to 293 cells can be detected, and that both the binding of virus to cells and infection can be blocked with antibodies specific for CD21. Two proteins known to form complexes with CD21 on the surface of lymphoid cells, CD35 and CD19, could not be detected at the surface of 293 cells. All infected clones of 293 cells exhibited tight latency with a pattern of gene expression similar to that of type II latency, but productive EBV replication and release of infectious virus could be induced inefficiently by forced expression of the lytic transactivators, R and Z. Low levels of mRNA specific for the transforming membrane protein of EBV, LMP-1, as well as for LMP-2, were detected; however, LMP-1 protein was either undetectable or near the limit of detection at less than 5% of the level typical of EBV-transformed B cells. A slight increase in expression of the receptor for epidermal growth factor, which can be induced in epithelial cells by LMP-1, was detected at the cell surface with two EBV-infected 293 cell clones. These results show that low levels of surface CD21 can support infection of an epithelial cell line by EBV. The results also raise the possibility that in a normal infection of epithelial cells by EBV, the LMP-1 protein is not expressed at levels that are high enough to be oncogenic and that there might be differences in the cells of EBV-associated epithelial cancers that have arisen to allow for elevated expression of LMP-1. Less
Matrix metalloproteinases (MMPs), in particular MMP-9, is induced by cytokines including IL-1 beta and contributes to airway injury and remodeling. However, the mechanism... More
Matrix metalloproteinases (MMPs), in particular MMP-9, is induced by cytokines including IL-1 beta and contributes to airway injury and remodeling. However, the mechanisms underlying IL-1 beta-induced MMP-9 expression and cell migration in human A549 cells remain unclear. Here, we report that the IL-1 beta-induced MMP-9 gene expression was mediated through the activation of p42/p44 MAPK, p38 MAPK, and JNK1/2 in A549 cells, determined by zymographic, RT-PCR, and Western blotting. The involvement of MAPKs in the IL-1beta-induced responses was further ensured by transfection with siRNA of MEK1, p42, p38, or JNK2. Moreover, the IL-1 beta-induced MMP-9 gene expression was also mediated through the translocation of NF-kappaB (p65) into the nucleus and the degradation of I kappaB alpha. In addition, the IL-1 beta-induced c-Jun phosphorylation was reduced by pretreatment with U0126 or SP600125. IL-1 beta stimulated the transcriptional activity of wild-type MMP-9 promoter in A549 cells, which was inhibited by U0126, SB203580, SP600125, and helenalin. In contrast, IL-1 beta had no effect on the cells transfected with a NF-kappaB-mutated MMP-9 promoter construct, suggesting that NF-kappaB is required for this response. Finally, the IL-1 beta-induced MMP-9 expression led to cell migration which was attenuated by pretreatment with U0126, SB203580, SP600125, helenalin, or MMP-2/9 inhibitor. These results suggested that in A549 cells, the activation of p42/p44 MAPK, p38 MAPK, JNK1/2, NF-kappaB, and AP-1 are essential for the IL-1 beta-induced MMP-9 gene expression and cell migration. Less
Matrix metalloproteinases (MMPs), in particular MMP-9, have been shown to be induced by cytokines including tumor necrosis factor-alpha (TNF-alpha) and contributes to air... More
Matrix metalloproteinases (MMPs), in particular MMP-9, have been shown to be induced by cytokines including tumor necrosis factor-alpha (TNF-alpha) and contributes to airway inflammation. However, the mechanisms underlying MMP-9 expression induced by TNF-alpha in human A549 cells remain unclear. Here, we showed that TNF-alpha induced production of MMP-9 protein and mRNA is determined by zymographic, Western blotting, RT-PCR and ELISA assay, which were attenuated by inhibitors of MEK1/2 (U0126), JNK (SP600125), and NF-kappaB (helenalin), and transfection with dominant negative mutants of ERK2 (DeltaERK) and JNK (DeltaJNK), and siRNAs for MEK1, p42 and JNK2. TNF-alpha-stimulated phosphorylation of p42/p44 MAPK and JNK were attenuated by pretreatment with the inhibitors U0126 and SP600125 or transfection with dominant negative mutants of DeltaERK and DeltaJNK. Furthermore, the involvement of NF-kappaB in TNF-alpha-induced MMP-9 production was consistent with that TNF-alpha-stimulated degradation of IkappaB-alpha and translocation of NF-kappaB into the nucleus which were blocked by helenalin, but not by U0126 and SP600125, revealed by immunofluorescence staining. The regulation of MMP-9 gene transcription by MAPKs and NF-kappaB was further confirmed by gene luciferase activity assay. MMP-9 promoter activity was enhanced by TNF-alpha in A549 cells transfected with wild-type MMP-9-Luc, which was inhibited by helenalin, U0126, or SP600125. In contrast, TNF-alpha-stimulated MMP-9 luciferase activity was totally lost in cells transfected with mutant-NF-kappaB MMP-9-luc. Moreover, pretreatment with actinomycin D and cycloheximide attenuated TNF-alpha-induced MMP-9 expression. These results suggest that in A549 cells, phosphorylation of p42/p44 MAPK, JNK, and transactivation of NF-kappaB are essential for TNF-alpha-induced MMP-9 gene expression. Less
Francisella tularensis, an intracellular pathogen, is highly virulent when inhaled. Alveolar epithelial type I (ATI) and type II (ATII) cells line the majority of the alv... More
Francisella tularensis, an intracellular pathogen, is highly virulent when inhaled. Alveolar epithelial type I (ATI) and type II (ATII) cells line the majority of the alveolar surface and respond to inhaled pathogenic bacteria via cytokine secretion. We hypothesized that these cells contribute to the lung innate immune response to F. tularensis. Results demonstrated that the live vaccine strain (LVS) contacted ATI and ATII cells by 2 h following intranasal inoculation of mice. In culture, primary human ATI or ATII cells, grown on transwell filters, were stimulated on the apical (AP) surface with virulent F. tularensis Schu 4 or LVS. Basolateral (BL) conditioned medium (CM), collected 6 and 24 h later, was added to the BL surfaces of transwell cultures of primary human pulmonary microvasculature endothelial cells (HPMEC) prior to the addition of polymorphonuclear leukocytes (PMNs) or dendritic cells (DCs) to the AP surface. HPMEC responded to S4- or LVS-stimulated ATII, but not ATI, CM as evidenced by PMN and DC migration. Analysis of the AP and BL ATII CM revealed that both F. tularensis strains induced various levels of a variety of cytokines via NF-kappaB activation. ATII cells pretreated with an NF-kappaB inhibitor prior to F. tularensis stimulation substantially decreased interleukin-8 secretion, which did not occur through Toll-like receptor 2, 2/6, 4, or 5 stimulation. These data indicate a crucial role for ATII cells in the innate immune response to F. tularensis. Less
Severe acute respiratory syndrome coronavirus (SARS-CoV) emerged in 2002 as an important cause of severe lower respiratory tract infection in humans, and in vitro models ... More
Severe acute respiratory syndrome coronavirus (SARS-CoV) emerged in 2002 as an important cause of severe lower respiratory tract infection in humans, and in vitro models of the lung are needed to elucidate cellular targets and the consequences of viral infection. The SARS-CoV receptor, human angiotensin 1-converting enzyme 2 (hACE2), was detected in ciliated airway epithelial cells of human airway tissues derived from nasal or tracheobronchial regions, suggesting that SARS-CoV may infect the proximal airways. To assess infectivity in an in vitro model of human ciliated airway epithelia (HAE) derived from nasal and tracheobronchial airway regions, we generated recombinant SARS-CoV by deletion of open reading frame 7a/7b (ORF7a/7b) and insertion of the green fluorescent protein (GFP), resulting in SARS-CoV GFP. SARS-CoV GFP replicated to titers similar to those of wild-type viruses in cell lines. SARS-CoV specifically infected HAE via the apical surface and replicated to titers of 107 PFU/ml by 48 h postinfection. Polyclonal antisera directed against hACE2 blocked virus infection and replication, suggesting that hACE2 is the primary receptor for SARS-CoV infection of HAE. SARS-CoV structural proteins and virions localized to ciliated epithelial cells. Infection was highly cytolytic, as infected ciliated cells were necrotic and shed over time onto the luminal surface of the epithelium. SARS-CoV GFP also replicated to a lesser extent in ciliated cell cultures derived from hamster or rhesus monkey airways. Efficient SARS-CoV infection of ciliated cells in HAE provides a useful in vitro model of human lung origin to study characteristics of SARS-CoV replication and pathogenesis. Less
Cellular senescence is a state of irreversible growth arrest induced either by telomere shortening (replicative senescence) or by telomere-independent signals (stress-ind... More
Cellular senescence is a state of irreversible growth arrest induced either by telomere shortening (replicative senescence) or by telomere-independent signals (stress-induced senescence). The alveolar epithelium is often injured by a variety of inhaled toxins, including cigarette smoke (CS). In the present study, we investigated whether exposure to CS induces senescence of alveolar epithelial cells. In vitro experiments showed that exposure of A549 cells or normal human alveolar epithelial cells to sublethal concentrations of aqueous CS extracts induced cellular senescence. The senescence was characterized by a dose- and time-dependent increase in senescence-associated beta-galactosidase activity, senescence-associated changes in cell morphology, an increase in cell size and lysosomal mass, accumulation of lipofuscin, overexpression of p21(CIP1/WAF1/Sdi1) protein, and irreversible growth arrest. In vivo experiments in Institute for Cancer Research mice showed that inhalation of CS for 2 wk induced increases in senescence-associated beta-galactosidase activity, lipofuscin accumulation, and p21(CIP1/WAF1/Sdi1) protein expression in alveolar epithelial cells. These results suggest that CS induces a phenotype that is indistinguishable from that of senescence in alveolar epithelial cells. The induction of cellular senescence by CS may contribute to impaired re-epithelialization, leading to CS-related chronic lung diseases. Less
,-1-mg-ml--2.jpg)