Immunometabolic reprogramming is increasingly recognized as a driver of asthma pathogenesis, yet the molecular mechanisms linking lactate accumulation to airway inflammat... More
Immunometabolic reprogramming is increasingly recognized as a driver of asthma pathogenesis, yet the molecular mechanisms linking lactate accumulation to airway inflammation via protein lactylation (Kla) remain elusive. In this study, we integrated a house dust mite (HDM)-induced asthma model with quantitative lactylomics to identify ATP6V1B2, a key V-ATPase subunit, as a core lactylation target. Combined molecular dynamics simulations and biochemical analyses revealed that intracellular l-lactate triggers lactylation at K108/K109. This modification restricts ATP6V1B2 conformational flexibility, leading to the disassembly of the V1–V0 complex and subsequent loss of proton pump activity. Crucially, the lactylation event was validated in primary human bronchial epithelial cells (HBEs), confirming that HDM and l-lactate stimulation induce ATP6V1B2 lactylation, thereby ensuring the clinical relevance of our findings. We demonstrate that this loss-of-function precipitates lysosomal alkalinization and membrane permeabilization (LMP). Crucially, LMP acts as a central node that bifurcates into two pathogenic cascades: it triggers a catastrophic mitochondrial ROS burst via Cathepsin B leakage. This oxidative burst functions as a pivotal redox signal that initiates a non-canonical Caspase-8/3/GSDME-dependent pyroptosis pathway, distinct from intrinsic apoptosis. In vivo, blocking ATP6V1B2 lactylation using an AAV-delivered lactylation-deficient (2 KR) mutant successfully severed this metabolic-inflammatory loop, significantly attenuating airway inflammation, Th2 cytokine release, and tissue pyroptosis. These findings characterize a novel "l-lactate–ATP6V1B2–GSDME" axis, establishing ATP6V1B2 lactylation as a critical metabolic switch connecting lysosomal damage to inflammatory cell death, thereby identifying a potential therapeutic target for metabolic dysregulation in chronic asthma with severe pathology. Less
Aberrant activation of the hedgehog (Hh) signaling pathway has been implicated in the epithelial-to-mesenchymal transition (EMT) and cancer stem-like cell (CSC) maintenan... More
Aberrant activation of the hedgehog (Hh) signaling pathway has been implicated in the epithelial-to-mesenchymal transition (EMT) and cancer stem-like cell (CSC) maintenance; both processes can result in tumor progression and treatment resistance in several types of human cancer. Hh cooperates with the epidermal growth factor receptor (EGFR) signaling pathway in embryogenesis. We found that the Hh signaling pathway was silenced in EGFRTKI-sensitive non-small-cell lung cancer (NSCLC) cells, while it was inappropriately activated in EGFR-TKI-resistant NSCLC cells, accompanied by EMT induction and ABCG2 overexpression. Upregulation of Hh signaling through extrinsic SHH exposure downregulated E-cadherin expression and elevated Snail and ABCG2 expression, resulting in gefitinib tolerance (P < 0.001) in EGFR-TKI-sensitive cells. Blockade of the Hh signaling pathway using the SMO antagonist SANT-1 restored E-cadherin expression and downregulate Snail and ABCG2 in EGFR-TKI-resistant cells. A combination of SANT-1 and gefitinib markedly inhibited tumorigenesis and proliferation in EGFR-TKI-resistant cells (P < 0.001). These findings indicate that hyperactivity of Hh signaling resulted in EGFR-TKI resistance, by EMT introduction and ABCG2 upregulation, and blockade of Hh signaling synergistically increased sensitivity to EGFR-TKIs in primary and secondary resistant NSCLC cells. E-cadherin expression may be a potential biomarker of the suitability of the combined application of an Hh inhibitor and EGFR-TKIs in EGFR-TKI-resistant NSCLCs. Less
Polyhedral oligomeric silsesquioxane poly(carbonate-urea) urethane (POSS-PCU) is a versatile nanocomposite biomaterial with growing applications as a bioscaffold for tiss... More
Polyhedral oligomeric silsesquioxane poly(carbonate-urea) urethane (POSS-PCU) is a versatile nanocomposite biomaterial with growing applications as a bioscaffold for tissue engineering. Integration of synthetic implants with host tissue can be problematic but could be improved by topographical modifications. We describe optimization of POSS-PCU by dispersion of porogens (sodium bicarbonate (NaHCO3), sodium chloride (NaCl) and sucrose) onto the material surface, with the principle aim of increasing surface porosity, thus providing additional opportunities for improved cellular and vascular ingrowth. We assess the effect of the porogens on the material's mechanical strength, surface chemistry, wettability and cytocompatibilty. Surface porosity was characterized by scanning electron microscopy (SEM). There was no alteration in surface chemistry and wettability and only modest changes in mechanical properties were detected. The size of porogens correlated well with the porosity of the construct produced and larger porogens improved interconnectivity of spaces within constructs. Using primary human bronchial epithelial cells (HBECs) we demonstrate moderate in vitro cytocompatibility for all surface modifications; however, larger pores resulted in cellular aggregation. These cells were able to differentiate on POSS-PCU scaffolds. Implantation of the scaffold in vivo demonstrated that larger pore sizes favor cellular integration and vascular ingrowth. These experiments demonstrate that surface modification with large porogens can improve POSS-PCU nanocomposite scaffold integration and suggest the need to strike a balance between the non-porous surfaces required for epithelial coverage and the porous structure required for integration and vascularization of synthetic scaffolds in future construct design. Keywords: Biocompatible materials; Nanocomposites; Porosity; Re-epithelialization; Tissue engineering; Trachea. Copyright © 2016 The Authors. Published by Elsevier Ltd.. All rights reserved. Less
Cullin 4A (Cul4A) has been observed to be overexpressed in various cancers. In this study, the role of Cul4A in the growth and chemosensitivity in lung cancer cells were ... More
Cullin 4A (Cul4A) has been observed to be overexpressed in various cancers. In this study, the role of Cul4A in the growth and chemosensitivity in lung cancer cells were studied. We showed that Cul4A is overexpressed in lung cancer cells and tissues. Knockdown of the Cul4A expression by shRNA in lung cancer cells resulted in decreased cellular proliferation and growth in lung cancer cells. Increased sensitivity to gemcitabine, a chemotherapy drug, was also noted in those Cul4A knockdown lung cancer cells. Moreover, increased expression of p21, transforming growth factor (TGF)‐β inducible early gene‐1 (TIEG1) and TGF beta‐induced (TGFBI) was observed in lung cancer cells after Cul4A knockdown, which may be partially related to increased chemosensitivity to gemcitabine. G0/G1 cell cycle arrest was also noted after Cul4A knockdown. Notably, decreased tumour growth and increased chemosensitivity to gemcitabine were also noted after Cul4A knockdown in lung cancer xenograft nude mice models. In summary, our study showed that targeting Cul4A with RNAi or other techniques may provide a possible insight to the development of lung cancer therapy in the future. Less
P2Y receptor activation causes the release of inflammatory cytokines in the bronchial epithelium, whereas G protein-coupled estrogen receptor (GPER), a novel estrogen (E2... More
P2Y receptor activation causes the release of inflammatory cytokines in the bronchial epithelium, whereas G protein-coupled estrogen receptor (GPER), a novel estrogen (E2) receptor, may play an anti-inflammatory role in this process. We investigated the cellular mechanisms underlying the inhibitory effect of GPER activation on the P2Y receptor-mediated Ca2+ signaling pathway and cytokine production in airway epithelia. Expression of GPER in primary human bronchial epithelial (HBE) or 16HBE14o- cells was confirmed on both the mRNA and protein levels. Stimulation of HBE or 16HBE14o- cells with E2 or G1, a specific agonist of GPER, attenuated the nucleotide-evoked increases in [Ca2+]i, whereas this effect was reversed by G15, a GPER-specific antagonist. G1 inhibited the secretion of two proinflammatory cytokines, interleukin (IL)-6 and IL-8, in cells stimulated by adenosine 5′-(γ-thio)triphosphate (ATPγS). G1 stimulated a real-time increase in cAMP levels in 16HBE14o- cells, which could be inhibited by adenylyl cyclase inhibitors. The inhibitory effects of E2 or G1 on P2Y receptor-induced increases in Ca2+ were reversed by treating the cells with a protein kinase A (PKA) inhibitor. These results demonstrated that the inhibitory effects of G1 or E2 on P2Y receptor-mediated Ca2+ mobilization and cytokine secretion were due to GPER-mediated activation of a cAMP-dependent PKA pathway. This study has reported, for the first time, the expression and function of GPER as an anti-inflammatory component in human bronchial epithelia, which may mediate through its opposing effects on the pro‐inflammatory pathway activated by the P2Y receptors in inflamed airway epithelia. Less
Autophagy, a type II programmed cell death, is essential for cell survival under stress, e.g. lung injury, and bone marrow-derived mesenchymal stem cells (BM-MSCs) have g... More
Autophagy, a type II programmed cell death, is essential for cell survival under stress, e.g. lung injury, and bone marrow-derived mesenchymal stem cells (BM-MSCs) have great potential for cell therapy. However, the mechanisms underlying the BM-MSC activation of autophagy to provide a therapeutic effect in ischaemia/reperfusion-induced lung injury (IRI) remain unclear. Thus, we investigate the activation of autophagy in IRI following transplantation with BM-MSCs. Seventy mice were pre-treated with BM-MSCs before they underwent lung IRI surgery in vivo. Human pulmonary micro-vascular endothelial cells (HPMVECs) were pre-conditioned with BM-MSCs by oxygen-glucose deprivation/reoxygenation (OGD) in vitro. Expression markers for autophagy and the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) signalling pathway were analysed. In IRI-treated mice, administration of BM-MSCs significantly attenuated lung injury and inflammation, and increased the level of autophagy. In OGD-treated HPMVECs, co-culture with BM-MSCs attenuated endothelial permeability by decreasing the level of cell death and enhanced autophagic activation. Moreover, administration of BM-MSCs decreased the level of PI3K class I and p-Akt while the expression of PI3K class III was increased. Finally, BM-MSCs-induced autophagic activity was prevented using the inhibitor LY294002. Administration of BM-MSCs attenuated lung injury by improving the autophagy level via the PI3K/Akt signalling pathway. These findings provide further understanding of the mechanisms related to BM-MSCs and will help to develop new cell-based therapeutic strategies in lung injury. Less
TRIM protein family is an evolutionarily conserved gene family implicated in a number of
Background: Protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme capable of methylating histone and non-histone proteins, and is involved in a wid... More
Background: Protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme capable of methylating histone and non-histone proteins, and is involved in a wide range of cellular processes that range from transcriptional regulation to organelle biosynthesis. As such, its overexpression has been linked to tumor suppressor gene silencing, enhanced tumor cell growth and survival. Material and methods: Quantitative real-time polymerase chain reaction, Western immunoblot and immunohistochemistry were used to characterize PRMT5 expression in lung cancer cell lines and human tumors. Clinicopathological findings of tissue microarray based samples from 229 patients with non-small cell lung carcinomas (NSCLC) and 133 cases with pulmonary neuroendocrine tumors (NET) were analyzed with regard to nuclear and cytoplasmic PRMT5 expression. Results: There was statistically significant difference in PRMT5 messenger RNA expression between tumors and nonneoplastic lung tissues. Immunoblot experiments showed abundant expression of PRMT5 and its symmetric methylation mark H4R3 in lung carcinoma but not in non-neoplastic human pulmonary alveolar and bronchial epithelial cell lines. More than two thirds of lung tumors expressed PRMT5. High levels of cytoplasmic PRMT5 were detected in 20.5% of NSCLC and in 16.5% of NET; high levels of nuclear PRMT5 were detected in 38.0% of NSCLC and 24.0% of NET. Cytoplasmic PRMT5 was associated with high grade in both NSCLC and pulmonary NET while nuclear PRMT5 was more frequent in carcinoid tumors (p < 0.05). Conclusion: The observed findings support the role of PRMT5 in lung tumorigenesis and reflect its functional dichotomy in cellular compartments. Virtual slide: The virtual slides for this article can be found here: http://www.diagnosticpathology.diagnomx.eu/vs/1611895162102528. Less
Background: Protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme capable of methylating histone and non-histone proteins, and is involved in a wid... More
Background: Protein arginine methyltransferase-5 (PRMT5) is a chromatin-modifying enzyme capable of methylating histone and non-histone proteins, and is involved in a wide range of cellular processes that range from transcriptional regulation to organelle biosynthesis. As such, its overexpression has been linked to tumor suppressor gene silencing, enhanced tumor cell growth and survival. Material and methods: Quantitative real-time polymerase chain reaction, Western immunoblot and immunohistochemistry were used to characterize PRMT5 expression in lung cancer cell lines and human tumors. Clinicopathological findings of tissue microarray based samples from 229 patients with non-small cell lung carcinomas (NSCLC) and 133 cases with pulmonary neuroendocrine tumors (NET) were analyzed with regard to nuclear and cytoplasmic PRMT5 expression. Results: There was statistically significant difference in PRMT5 messenger RNA expression between tumors and nonneoplastic lung tissues. Immunoblot experiments showed abundant expression of PRMT5 and its symmetric methylation mark H4R3 in lung carcinoma but not in non-neoplastic human pulmonary alveolar and bronchial epithelial cell lines. More than two thirds of lung tumors expressed PRMT5. High levels of cytoplasmic PRMT5 were detected in 20.5% of NSCLC and in 16.5% of NET; high levels of nuclear PRMT5 were detected in 38.0% of NSCLC and 24.0% of NET. Cytoplasmic PRMT5 was associated with high grade in both NSCLC and pulmonary NET while nuclear PRMT5 was more frequent in carcinoid tumors (p < 0.05). Conclusion: The observed findings support the role of PRMT5 in lung tumorigenesis and reflect its functional dichotomy in cellular compartments. Virtual slide: The virtual slides for this article can be found here: http://www.diagnosticpathology.diagnomx.eu/vs/1611895162102528. Less
Tumor cells display a different profile of gene expression than their normal counterparts. Perturbations in the levels of cellular splicing factors can alter gene express... More
Tumor cells display a different profile of gene expression than their normal counterparts. Perturbations in the levels of cellular splicing factors can alter gene expression, potentially leading to tumorigenesis. We found that splicing factor SRp20 (SFRS3) is highly expressed in cancers. SRp20 regulated the expression of Forkhead box transcription factor M1 (FoxM1) and two of its transcriptional targets, PLK1 and Cdc25B, and controlled cell cycle progression and proliferation. Cancer cells with RNAi-mediated reduction of SRp20 expression exhibited G2/M arrest, growth retardation, and apoptosis. Increased SRp20 expression in rodent fibroblasts promoted immortal cell growth and transformation. More importantly, we found that SRp20 promoted tumor induction and the maintenance of tumor growth in nude mice and rendered immortal rodent fibroblasts tumorigenic. Collectively, these results suggest that increased SRp20 expression in tumor cells is a critical step for tumor initiation, progression, and maintenance. Keywords: Cancer; G2/M arrest; SFRS3; SRp20; cell transformation; splicing factors; tumor induction. Less
The transcription factor hypoxia-inducible factor (HIF)-1 plays a central physiological role in oxygen and energy homeostasis, and is activated during hypoxia by stabiliz... More
The transcription factor hypoxia-inducible factor (HIF)-1 plays a central physiological role in oxygen and energy homeostasis, and is activated during hypoxia by stabilization of the subunit HIF-1α. Recent studies have demonstrated that non-hypoxic stimuli can also activate HIF-1α in a cell-specific manner. Here, we demonstrate that stimulation of BEAS-2B cells and primary human bronchial epithelial cells by proinflammatory cytokines TNFα/IL-4 strongly induced expression and transcriptional activity of HIF-1α under normoxic conditions and amplified hypoxic HIF-1α activation. TNFα/IL-4 stimulated de novo HIF-1α gene transcription and translation rather than affected HIF-1α protein degradation and mRNA decay process. The activation of HIF-1α by TNFα/IL-4 was countered by the phosphoinositol 3-kinase (PI3K) inhibitor LY-294002 and rapamycin, an antagonist of mammalian target of rapamycin (mTOR), but not by inhibition of the MAPK pathway. In line, TNFα/IL-4 also activated NF-κB, whereas blocking of NF-κB by an inhibitor or silencing NF-κB subunit p65 attenuated HIF-1α activation by TNFα/IL-4. We also found the collaborative induction of VEGF, a potent angiogenic factor required for airway remodeling, by TNFα/IL-4 and hypoxia partially via HIF-1α pathway in BEAS-2B cells. This study reports the previously unsuspected collaborative regulation of HIF-1α by TNFα/IL-4 and hypoxia in bronchial epithelial cells partially via PI3K-mTOR and NF-κB pathway, and thereby will lead to the elucidation of the importance of HIF-1 in integrating inflammatory and hypoxic response in the pathogenesis of airway diseases. hypoxia activates a number of genes that are important in the cellular adaptation to low oxygen conditions. Hypoxia inducible factor (HIF)-1 is a key regulator of hypoxia-inducible genes like erythropoietin and vascular endothelial growth factor (VEGF), as well as a growing number of glycolytic and metabolic enzymes (20). These genes are implicated in many different cellular functions such as cell survival, cell proliferation, apoptosis, glucose metabolism, and angiogenesis. The heterodimeric HIF-1 is composed of an oxygen-sensitive HIF-1α and a constitutive HIF-1β subunit, which both belong to the family of basic helix-loop-helix and PAS domain proteins (6, 34). HIF-1 activity is primarily regulated by the abundance of the HIF-1α subunit. Under normoxic conditions, HIF-1α is targeted for rapid degradation by hydroxylation of specific prolyl residues (Pro 402 and Pro 564), which are catalyzed by specific oxygenases, identified as prolyhydroxylase domain-containing proteins (PHDs) (43). Another factor in the degradation process is the Von Hippel-Lindau tumor suppressor protein, which facilitates degradation of HIF-1α through the ubiquitin-proteasome pathway(30). Under hypoxic conditions, HIF-1α is stabilized, translocates into the nucleus where it dimerizes with HIF-1β, and transactivates downstream target genes containing hypoxia-response elements (HRE) within their promoter or enhancer. In addition to hypoxia, growth factors, lipopolysaccharides, and proinflammatory cytokines such as IL-1β and TNFα have been demonstrated to be critical regulators of HIF-1α activation(3, 13, 19, 50). There is growing evidence that HIF-1α is involved in the inflammatory process by regulating angiogenesis and functions of inflammatory cells (11, 53). Persistent inflammation of the respiratory tract in chronic pulmonary disease is mediated by increased expression of multiple inflammation mediators (1). The bronchial airway epithelium plays a critical role not only in the maintenance of physicochemical homoeostasis of the airways but also in the pathogenesis of airway diseases. During airway inflammation the epithelium is both a source of mediator production as well as a target of remodeling processes(42). Immunohistochemistry revealed induction of HIF-1α in bronchial epithelium cells by hypoxia (51). Studies showed that HIF-1α play an intergrative role in conditions of hypoxia and inflammation, and the microenvironments of bronchial epithelial cells are characterized by inflammatory and hypoxic conditions (39). Moreover, an orchestrated change among posttranscriptional mechanisms in human bronchial epithelial BEAS-2B cells following inflammatory stimulation has been documented, and treatment of BEAS-2B cells with IL-4/TNFα can increase the expression of a number of genes coding for cytokines and chemokines (52). Thus, in the present study, we investigated the potential activation of HIF-1α by IL-4/TNFα and hypoxia in bronchial airway epithelium cells and identified the signaling pathways involved in the process. We found that TNFα/IL-4 induced HIF-1α transcription and translation in BEAS-2B cells and primary human bronchial epithelial cells under normoxic conditions and amplified hypoxic HIF-1α activation. This response involves the activation of the phosphatidylinositol 3-kinase (PI3K)-mammalian target of rapamycin (mTOR) pathway and of NF-κB. Furthermore, TNFα/IL-4 and hypoxia induced VEGF expression synergistically via the HIF-1α pathway. Because the microenvironments of bronchial epithelial cells are characterized by inflammatory and hypoxic conditions during the pathogenesis of inflammatory airway diseases, the cooperative regulation of HIF-1α by inflammatory cytokines and hypoxia provides important new insight into inflammatory airway diseases and identifies novel therapeutic targets. Less
Basophils are the accessory cell type for T-helper (Th)2 induction and initiators in immunoglobulin E-mediated chronic allergic inflammation. Basophils and Th17 cells acc... More
Basophils are the accessory cell type for T-helper (Th)2 induction and initiators in immunoglobulin E-mediated chronic allergic inflammation. Basophils and Th17 cells accumulate at the inflammatory sites, such as the airways of allergic asthmatic patients. We investigated the activation of interleukin (IL)-17A on the primary human basophils/KU812 basophilic cells and primary human bronchial epithelial cells/BEAS-2B bronchial epithelial cells. Cytokines, chemokines, adhesion molecules and intracellular signalling molecules were assayed by ELISA or flow cytometry. Co-culture of bronchial epithelial cells and basophils could significantly induce the release of IL-6, an epithelial inflammatory cytokine, and CCL2, a chemokine for basophils, esosinophils and monocytes. Such induction was synergistically enhanced by IL-17A, and direct interaction between these two cells was necessary for IL-17A-induced IL-6 and CCL2 release. Surface expression of intercellular adhesion molecule-1 on bronchial epithelial cells was also upregulated upon their interaction. The interaction of basophils and bronchial epithelial cells under IL-17A stimulation was differentially regulated by extracellular signal-regulated kinase, c-Jun N-terminal protein kinase, p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways. These findings suggest a novel immunopathological role of Th17 cells and basophils in allergic asthma through the activation of granulocyte-mediated inflammation initiated by the direct interaction between basophils and bronchial epithelial cells. Less
Lung cancer is the most frequent cause of cancer-related death in this country for men and women. MicroRNAs (miRNAs) are a family of small non-coding RNAs (approximately ... More
Lung cancer is the most frequent cause of cancer-related death in this country for men and women. MicroRNAs (miRNAs) are a family of small non-coding RNAs (approximately 21–25 nt long) capable of targeting genes for either degradation of mRNA or inhibition of translation. We identified aberrant expression of 41 miRNAs in lung tumor versus uninvolved tissue. MiR-133B had the lowest expression of miRNA in lung tumor tissue (28 fold reduction) compared to adjacent uninvolved tissue. We identified two members of the BCL-2 family of pro-survival molecules (MCL-1 and BCL2L2 (BCLw)) as predicted targets of miR-133B. Selective over-expression of miR-133B in adenocarcinoma (H2009) cell lines resulted in reduced expression of both MCL-1 and BCL2L2. We then confirmed that miR-133B directly targets the 3’UTRs of both MCL-1 and BCL2L2. Lastly, over-expression of miR-133B induced apoptosis following gemcitabine exposure in these tumor cells. To our knowledge, this represents the first observation of decreased expression of miR-133B in lung cancer and that it functionally targets members of the BCL-2 family. Less
Primary biliary cirrhosis (PBC) is characterized by antimitochondrial antibodies (AMA), directed to the E2 component of the pyruvate dehydrogenase complex (PDC-E2). Notwi... More
Primary biliary cirrhosis (PBC) is characterized by antimitochondrial antibodies (AMA), directed to the E2 component of the pyruvate dehydrogenase complex (PDC-E2). Notwithstanding the presence of mitochondria in virtually all nucleated cells, the destruction in PBC is limited to small intrahepatic bile ducts. The reasons for this tissue specificity remain unknown, although biliary epithelial cells (BEC) uniquely preserve the PDC-E2 epitope following apoptosis. Notably, PBC recurs in an allogeneic transplanted liver, suggesting generic rather than host-PBC-specific susceptibility of BEC. We used cultured human intrahepatic BEC (HIBEC) and other well-characterized cell lines, including, HeLa, CaCo-2 cells, and non transformed human keratinocytes and bronchial epithelial cells (BrEpC), to determine the integrity and specific localization of PDC-E2 during induced apoptosis. All cell lines, both before and after apoptosis, were tested with sera from patients with PBC (n=30), other autoimmune liver and rheumatic diseases (n=20), and healthy individuals (n=20), a mouse monoclonal antibody against PDC-E2, and AMA with an IgA isotype. PDC-E2 was found to localize unmodified within apoptotic blebs of HIBEC, but not within blebs of various other cell lineages studied. The fact that AMA- containing sera reacted with PDC-E2 on apoptotic BEC without a requirement for permeabilization suggests that the autoantigen is accessible to the immune system during apoptosis. In conclusion, our data indicate that the tissue (cholangiocyte) specificity of the autoimmune injury in PBC is a consequence of the unique characteristics of HIBEC during apoptosis and can be explained by exposure to the immune system of intact immunoreactive PDC-E2 within apoptotic blebs. Keywords: autoimmunity, antimitochondrial antibodies, apoptosis, apoptotic bodies, cell clearance Less
Crk is a member of a family of adaptor proteins that are involved in intracellular signal pathways altering cell adhesion, proliferation, and migration. Increased express... More
Crk is a member of a family of adaptor proteins that are involved in intracellular signal pathways altering cell adhesion, proliferation, and migration. Increased expression of Crk has been described in lung cancer and associated with increased tumor invasiveness. MicroRNAs (miRNAs) are a family of small non-coding RNAs (approximately 21-25 nt long) that are capable of targeting genes for either degradation of mRNA or inhibition of translation. Crk is a predicted putative target gene for miR-126. Over-expression of miR126 in a lung cancer cell line resulted in a decrease in Crk protein without any alteration in the associated mRNA. These lung cancer cells exhibit a decrease in adhesion, migration, and invasion. Decreased cancer cell invasion was also evident following targeted knockdown of Crk. MiR-126 alters lung cancer cell phenotype by inhibiting adhesion, migration, and invasion and the effects on invasion may be partially mediated through Crk regulation. Less
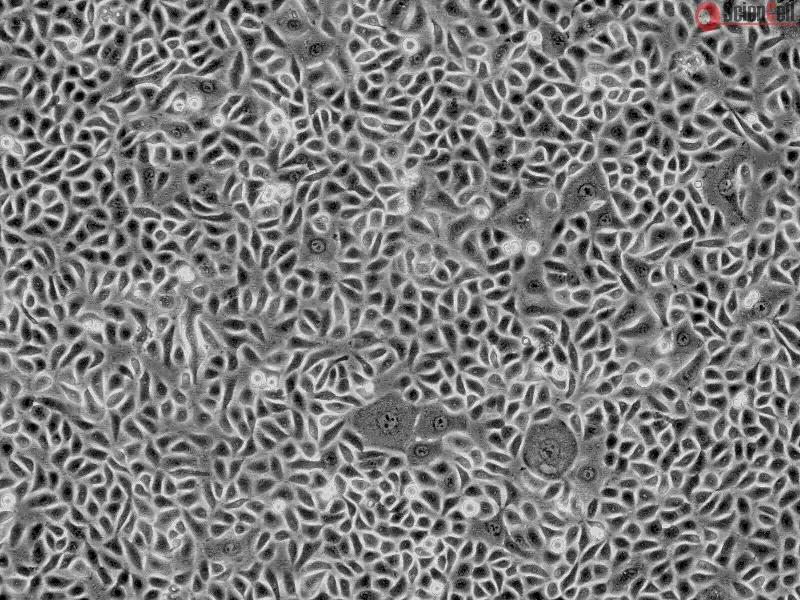
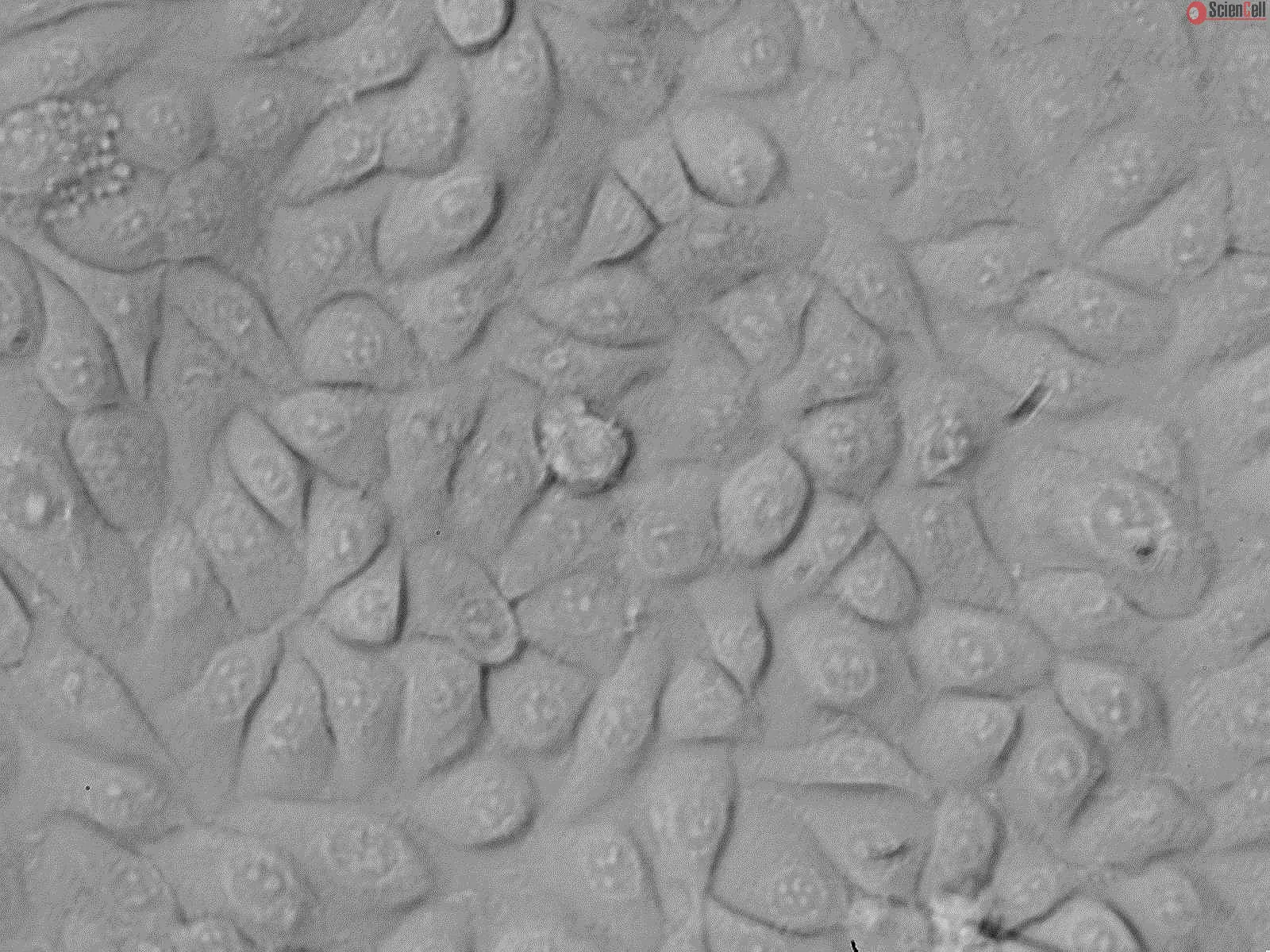




,-1-mg-ml--2.jpg)