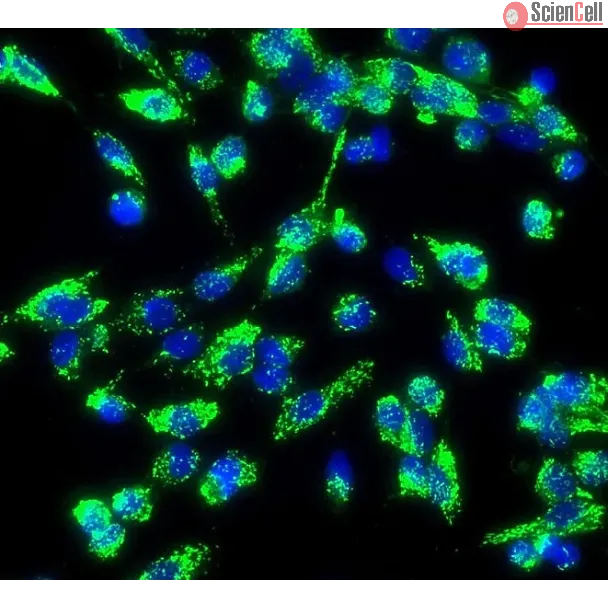
Emerging evidence has suggested the critical role of endothelial to mesenchymal transition (EndMT) in fibrotic diseases. The present study was designed to examine whether... More
Emerging evidence has suggested the critical role of endothelial to mesenchymal transition (EndMT) in fibrotic diseases. The present study was designed to examine whether EndMT is involved in arsenic trioxide (As2O3)-induced cardiac fibrosis and to explore the underlying mechanisms. Cardiac dysfunction was observed in rats after exposure to As2O3 for 15 days using echocardiography, and the deposition of collagen was detected by Masson’s trichrome staining and electron microscope. EndMT was indicated by the loss of endothelial cell markers (VE-cadherin and CD31) and the acquisition of mesenchymal cell markers (α-SMA and FSP1) determined by RT-PCR at the mRNA level and Western blot and immunofluorescence analysis at the protein level. In the in-vitro experiments, endothelial cells acquired a spindle-shaped morphology accompanying downregulation of the endothelial cell markers and upregulation of the mesenchymal cell markers when exposed to As2O3. As2O3 activated the AKT/GSK-3β/Snail signaling pathway, and blocking this pathway with PI3K inhibitor (LY294002) abolished EndMT in As2O3-treated endothelial cells. Our results highlight that As2O3 is an EndMT-promoting factor during cardiac fibrosis, suggesting that targeting EndMT is beneficial for preventing As2O3-induced cardiac toxicity. Less
Background Previous studies have shown that high glucose (HG) induced endothelial cell (EC) damage via a phenotypic transition of EC. There is increasing evidence suggest... More
Background Previous studies have shown that high glucose (HG) induced endothelial cell (EC) damage via a phenotypic transition of EC. There is increasing evidence suggesting the role of inflammatory cytokines in mediated HG-induced EC damage. However, little is known about the potential role of interleukin-1β (IL-1β) in the process. The aim of present study was to investigate whether IL-1β mediated HG–induced phenotypic transition in human aortic endothelial cells (HAECs) and to determine the possible underlying mechanism. Methods Primary HAECs were exposed to normal glucose (NG, 5.5 nM), high glucose (HG,30 nM), IL-1β (10 ng/ml), HG + IL-1β (10 ng/ml) and HG + anti-IL-1β antibodies (1000 ng/ml) or HG + IL-1β small interfering RNA (siRNA). Pathological changes were investigated using confocal microscopy and electron microscopy. Confocal microscopy was performed to detect the co-expression of CD31 and fibroblast specific protein 1 (FSP1). To study the effect of protein kinase C-β (PKCβ) activation on IL-1β in HAECs, HAECs were stimulated with 30 nM PMA (PKCβ activator) and 0.3 μM PKCβ inhibition (LY317615) for 48 h in the NG or HG group. The expressions of PKCβ and IL-1β were detected by RT-PCR and Western blot. And the concentration of IL-1β in the supernatant of HAECs was measured by ELISA. The expressions of FSP1, a-SMA and CD31 were detected by Western blot. Results It was shown that the HG resulted in significant increase in the expressions of PKCβ and IL-1β in dose-and time-dependent manners. The HG or exogenous IL-1β alone inhibited the expression of CD31 and markly increased the expressions of FSP1 and α-SMA. Furthermore, we observed that the HG and IL-1β synergistically increased FSP1 and a-SMA expressions compared with the HG or IL-1β alone group (P < 0.05). Confocal microscopy revealed a colocalization of CD31 and FSP1 and that some cells acquired spindle-shaped morphologies and a loss of CD31 staining. Electron microscopy showed that the HG resulted in the increased microfilamentation and a roughened endoplasmic reticulum structure in the cytoplasm. However, the changes above were attenuated by the intervention of anti-IL-1β antibodies or IL-1β siRNA (P < 0.05). In addition, the PMA induced the expressions of PKCβ and IL-1β in HAECs. The PKCβ activation may mediate the effect of the HG on IL-1β production, which could be attenuated by the PKCβ selective inhibitor (LY317615) (P < 0.05). Conclusions Our findings suggested that HG-induced phenotypic transition of HAECs might require IL-β activation via the PKCβ pathway. Less
Jujuboside B has been reported to have protective effect on many cardiovascular diseases. However, the effects of Jujuboside B on vascular tension and endothelial functio... More
Jujuboside B has been reported to have protective effect on many cardiovascular diseases. However, the effects of Jujuboside B on vascular tension and endothelial function are unknown. The present study investigated the effects of Jujuboside B on reducing vascular tension, protecting endothelial function and the potential mechanisms. The tension of isolated rat thoracic aorta ring was measured by Wire myograph system. The concentration of nitric oxide (NO) and the activity of endothelial nitric oxide synthase (eNOS) in human aortic endothelial cells (HAECs) were determined by Griess reagent method and enzyme-linked immune sorbent assay. The protein levels of eNOS and p-eNOS at Serine-1177 were determined by western blot analysis. Intracellular Ca2+ concentration in HAECs was measured by laser confocal imaging microscopy. Results showed that Jujuboside B reduced the tension of rat thoracic aorta rings with intact endothelium in a dose-dependent manner. L-NAME, KN93, EGTA, SKF96365, iberiotoxin and glibenclamide significantly attenuated Jujuboside B-induced vasodilation in endothelium-intact tissues. In contrast, indometacin and 4-DAMP had no such effects. Jujuboside B also promoted NO generation and increased eNOS activity, which were attenuated by L-NAME, EGTA and SKF96365. Moreover, Jujuboside B increased intracellular Ca2+ concentration dose-dependently, which was inhibited by EGTA and SKF96365. Besides, Jujuboside B induced a rapid Ca2+ influx instantaneously after depleting intracellular Ca2+ store, which was significantly inhibited by SKF96365. In conclusion, this study preliminarily confirmed that Jujuboside B reduced vascular tension endothelium-dependently. The underlying mechanisms involved that Jujuboside B increased extracellular Ca2+ influx through endothelial transient receptor potential cation (TRPC) channels, phosphorylated eNOS and promoted NO generation in vascular endothelial cells. In addition, Jujuboside B-induced vasodilation involved endothelium-dependent hyperpolarizaiton through endothelial potassium channels. Jujuboside B is a natural compound with new pharmacological effects on improving endothelial dysfunction and treating vascular diseases. Less
Autophagy, a type II programmed cell death, is essential for cell survival under stress, e.g. lung injury, and bone marrow-derived mesenchymal stem cells (BM-MSCs) have g... More
Autophagy, a type II programmed cell death, is essential for cell survival under stress, e.g. lung injury, and bone marrow-derived mesenchymal stem cells (BM-MSCs) have great potential for cell therapy. However, the mechanisms underlying the BM-MSC activation of autophagy to provide a therapeutic effect in ischaemia/reperfusion-induced lung injury (IRI) remain unclear. Thus, we investigate the activation of autophagy in IRI following transplantation with BM-MSCs. Seventy mice were pre-treated with BM-MSCs before they underwent lung IRI surgery in vivo. Human pulmonary micro-vascular endothelial cells (HPMVECs) were pre-conditioned with BM-MSCs by oxygen-glucose deprivation/reoxygenation (OGD) in vitro. Expression markers for autophagy and the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) signalling pathway were analysed. In IRI-treated mice, administration of BM-MSCs significantly attenuated lung injury and inflammation, and increased the level of autophagy. In OGD-treated HPMVECs, co-culture with BM-MSCs attenuated endothelial permeability by decreasing the level of cell death and enhanced autophagic activation. Moreover, administration of BM-MSCs decreased the level of PI3K class I and p-Akt while the expression of PI3K class III was increased. Finally, BM-MSCs-induced autophagic activity was prevented using the inhibitor LY294002. Administration of BM-MSCs attenuated lung injury by improving the autophagy level via the PI3K/Akt signalling pathway. These findings provide further understanding of the mechanisms related to BM-MSCs and will help to develop new cell-based therapeutic strategies in lung injury. Less
Background Human immunodeficiency virus (HIV) infection leads to decreased reverse cholesterol transport (RCT) in macrophages, and Nef mediated down-regulation and redist... More
Background Human immunodeficiency virus (HIV) infection leads to decreased reverse cholesterol transport (RCT) in macrophages, and Nef mediated down-regulation and redistribution of ATP-binding cassette transporter A1 (ABCA1) are identified as key factors for this effect. This may partially explain the increased risk of atherosclerosis in HIV infected individuals. Since endothelial dysfunction is key in the initial stages of atherosclerosis, we sought to determine whether RCT was affected in human aortic endothelial cells (HAECs). Results We found that apoA-I does not significantly stimulate cholesterol efflux in HAECs while cholesterol efflux to high-density lipoprotein (HDL) was dramatically reduced in HAECs co-cultured with HIV infected cells. Studies with wild type and Nef defective HIV revealed no significant differences suggesting that multiple factors are working perhaps in concert with Nef to affect cholesterol efflux to HDL from HAECs. Interestingly, treating HAECs with recombinant Nef showed similar effect in HDL mediated cholesterol efflux as observed in HAECs co-cultured with HIV infected cells. Using a detergent-free based subcellular fractionation approach, we demonstrated that exposure of HAECs to HIV infected cells or Nef alone disrupts caveolin 1 (Cav-1) subcellular trafficking upon HDL stimulation. Moreover, Nef significantly enhanced tyrosine 14 phosphorylation of Cav-1 which may have an impact on recycling of Cav-1 and caveolae. Conclusion These results suggest that HIV interferes with cholesterol efflux by HDL in HAECs through the disruption of Cav-1s’ cellular distribution and that multiple factors are involved, possibly including Nef, for the inhibition of HDL mediated cholesterol efflux and alteration of cellular distribution of Cav-1. Less
We investigated transcriptional control of gene expression in human abdominal aortic aneurysm (AAA). We previously identified 3274 differentially expressed genes in human... More
We investigated transcriptional control of gene expression in human abdominal aortic aneurysm (AAA). We previously identified 3274 differentially expressed genes in human AAA tissue compared to non-aneurysmal controls. Four expressed transcription factors (ELF1, ETS2, STAT5 and RUNX1) were selected for genome-wide chromatin immunoprecipitation. Transcription factor binding was enriched in 4760 distinct genes (FDR < 0.05), of which 713 were differentially expressed in AAA. Functional classification using Gene Ontology (GO), KEGG, and Network Analysis revealed enrichment in several biological processes including “leukocyte migration” (FDR = 3.09 × 10−05) and “intracellular protein kinase cascade” (FDR = 6.48 × 10−05). In the control aorta, the most significant GO categories differed from those in the AAA samples and included “cytoskeleton organization” (FDR = 1.24 × 10−06) and “small GTPase mediated signal transduction” (FDR = 1.24 × 10−06). Genes up-regulated in AAA tissue showed a highly significant enrichment for GO categories “leukocyte migration” (FDR = 1.62 × 10−11), “activation of immune response” (FDR = 8.44 × 10−11), “T cell activation” (FDR = 4.14 × 10−10) and “regulation of lymphocyte activation” (FDR = 2.45 × 10−09), whereas the down-regulated genes were enriched in GO categories “cytoskeleton organization” (FDR = 7.84 × 10−05), “muscle cell development” (FDR = 1.00 × 10−04), and “organ morphogenesis” (FDR = 3.00 × 10−04). Quantitative PCR assays confirmed a sub-set of the transcription factor binding sites including those in MTMR11, DUSP10, ITGAM, MARCH1, HDAC8, MMP14, MAGI1, THBD and SPOCK1. Less
Mitochondrial injury and dysfunction, a significant feature in metabolic syndrome, triggers endothelial cell dysfunction and cell death. Increasing evidence suggests that... More
Mitochondrial injury and dysfunction, a significant feature in metabolic syndrome, triggers endothelial cell dysfunction and cell death. Increasing evidence suggests that mitophagy, a process of autophagic turnover of damaged mitochondria, maintains mitochondrial integrity. PINK1 (phosphatase and tensin homolog (PTEN)-induced putative kinase 1) and Parkin signaling is a key pathway in mitophagy control. In this study, we examined whether this pathway could protect mitochondria under metabolic ... Less
Magnesium (Mg) based alloys are the most advanced cardiovascular stent materials. This new generation of stent scaffold is currently under clinical evaluation with encour... More
Magnesium (Mg) based alloys are the most advanced cardiovascular stent materials. This new generation of stent scaffold is currently under clinical evaluation with encouraging outcomes. All these Mg alloys contain a certain amount of rare earth (RE) elements though the exact composition is not yet disclosed. RE alloying can usually enhance the mechanical strength of different metal alloys but their toxicity might be an issue for medical applications. It is still unclear how RE elements will affect the magnesium (Mg) alloys intended for stent materials as a whole. In this study, we evaluated MgZnCaY-1RE, MgZnCaY-2RE, MgYZr-1RE, and MgZnYZr-1RE alloys for cardiovascular stents applications regarding their mechanical strength, corrosion resistance, hemolysis, platelet adhesion/activation, and endothelial biocompatibility. The mechanical properties of all alloys were significantly improved. Potentiodynamic polarization showed that the corrosion resistance of four alloys was at least 3–10 times higher than that of pure Mg control. Hemolysis test revealed that all the materials were non-hemolytic while little to moderate platelet adhesion was found on all materials surface. No significant cytotoxicity was observed in human aorta endothelial cells cultured with magnesium alloy extract solution for up to seven days. Direct endothelialization test showed that all the alloys possess significantly better capability to sustain endothelial cell attachment and growth. The results demonstrated the promising potential of these alloys for stent material applications in the future. Less
In professional immune cells, Toll-like receptor 4 (TLR4) induces tightly regulated inflammatory response to avoid tissue damage via the induction of "endotoxin tolerance... More
In professional immune cells, Toll-like receptor 4 (TLR4) induces tightly regulated inflammatory response to avoid tissue damage via the induction of "endotoxin tolerance", which is a transient state of cell desensitization in response to lipopolysaccharide (LPS) restimulation after a prior LPS exposure. However, in endothelial cells, the regulation of TLR4-induced inflammation is not fully understood. In this study, we found that the gene transcripts for a lot of Toll-like receptors were expressed in various endothelial cells, including human umbilical vein endothelial cells (HUVEC), human aortic endothelial cell (HAEC), and mouse microvascular endothelial cells (bEND.3). Proteins of TLR4 and its coreceptor CD14 were also detected in HUVEC. LPS treatment significantly upregulated the expression of proinflammation cytokines such as IL-1β, IL-6, and IL-8 only in HUVEC, but not in HAEC and bEND.3, suggesting that vein endothelial cells are important source of proinflammatory cytokines in response to LPS. Unexpectedly, "endotoxin tolerance" was not induced in endothelial cell, but was induced in control glial cells, as LPS pretreatment downregulated the cytokine expression in control glial cells, but did not in endothelial cells, when the cells were restimulated with LPS. The upregulation of cytokine gene expression was dependent on NF-κB signaling, and NF-κB inhibitor repressed the induction of cytokines. Two important signal molecules MyD88 and TRIF, which are TLR4 downstream and NF-κB upstream, were upregulated in vein endothelial cells but were downregulated in control glial cells. These results suggested that vein endothelial cells may play important roles in the pathophysiology of systemic inflammation-associated diseases such as sepsis and septic cardiomyopathy. Less
Autophagy is a cytoprotective pathway used to degrade and recycle cytoplasmic content. Dysfunctional autophagy has been linked to both cancer and cardiomyopathies. Here, ... More
Autophagy is a cytoprotective pathway used to degrade and recycle cytoplasmic content. Dysfunctional autophagy has been linked to both cancer and cardiomyopathies. Here, we show a role for the transcriptional regulator p8 in autophagy. p8 RNA interference (RNAi) increases basal autophagy markers in primary cardiomyocytes, in H9C2 and U2OS cells, and decreases cellular viability after autophagy induction. This autophagy is associated with caspase activation and is blocked by atg5 silencing and by pharmacological inhibitors. FoxO3 transcription factor was reported to activate autophagy by enhancing the expression of autophagy-related genes. P8 expression represses FoxO3 transcriptional activity, and p8 knockdown affects FoxO3 nuclear localization. Thus, p8 RNAi increases FoxO3 association with bnip3 promoter, a known proautophagic FoxO3 target, resulting in higher bnip3 RNA and protein levels. Accordingly, bnip3 knockdown restores cell viability and blocks apoptosis of p8-deficient cells. In vivo, p8 −/− mice have higher autophagy and express higher cardiac bnip3 levels. These mice develop left ventricular wall thinning and chamber dilation, with consequent impaired cardiac function. Our studies provide evidence of a p8-dependent mechanism regulating autophagy by acting as FoxO3 corepressor, which may be relevant for diseases associated with dysregulated autophagy, as cardiovascular pathologies and cancer. Less
Although the liver is known to be the main site of factor VIII (FVIII) production, other organs are probably also important for the regulation of FVIII secretion. However... More
Although the liver is known to be the main site of factor VIII (FVIII) production, other organs are probably also important for the regulation of FVIII secretion. However, the study of the regulation of extrahepatic FVIII production has been hampered by the lack of definitive identification of human tissues able to secrete FVIII. Recent studies have shown that lung endothelial cells can synthesize FVIII. We therefore studied the production of FVIII by endothelial cells purified from other vascular beds. Because physiologic stress results in a rapid elevation of FVIII, we also investigated whether endothelial cells can store FVIII and secrete it after treatment with agonists. Microvascular endothelial cells from lung, heart, intestine, and skin as well as endothelial cells from pulmonary artery constitutively secreted FVIII and released it after treatment with phorbol-myristate acetate and epinephrine. By contrast, endothelial cells from the aorta, umbilical artery and umbilical vein did not constitutively secrete FVIII or release it after treatment with agonists, probably because of a lack of FVIII synthesis. Extrahepatic endothelial cells from certain vascular beds therefore appear to be an important FVIII production and storage site with the potential to regulate FVIII secretion in chronic and acute conditions. Less
Angiogenesis and apoptosis are reciprocal processes in endothelial cells. Bcl-2, an anti-apoptotic protein, has been found to have angiogenic activities. The purpose of t... More
Angiogenesis and apoptosis are reciprocal processes in endothelial cells. Bcl-2, an anti-apoptotic protein, has been found to have angiogenic activities. The purpose of this study was to determine the role of Bcl-2 in hypoxia-induced angiogenesis in endothelial cells and to investigate the underlying mechanisms. Human aortic endothelial cells (HAECs) were exposed to hypoxia followed by reoxygenation. Myocardial ischemia and reperfusion mouse model was used and Bcl-2 expression was assessed. Bcl-2 expression increased in a time-dependent manner in response to hypoxia from 2 to 72h. Peak expression occurred at 12h (3- to 4-fold, p<0.05). p38 inhibitor (SB203580) blocked hypoxia-induced Bcl-2 expression, whereas PKC, ERK1/2 and PI3K inhibitors did not. Knockdown of Bcl-2 resulted in decreased HAECs' proliferation and migration. Over-expression of Bcl-2 increased HAECs' tubule formation, whereas knockdown of Bcl-2 inhibited this process. In this model of myocardial ischemia and reperfusion, Bcl-2 expression was increased and was associated with increased p38 MAPK activation. Our results showed that hypoxia induces Bcl-2 expression in HAECs via p38 MAPK pathway. Published by Elsevier Inc. Less
Advanced glycated end-product receptor 1 (AGER1) protects against vascular disease promoted by oxidants, such as advanced glycated end products (AGEs), via inhibition of ... More
Advanced glycated end-product receptor 1 (AGER1) protects against vascular disease promoted by oxidants, such as advanced glycated end products (AGEs), via inhibition of reactive oxygen species (ROS). However, the specific AGEs, sources, and pathways involved remain undefined. The mechanism of cellular NADPH oxidase (NOX)-dependent ROS generation by defined AGEs, N(epsilon)-carboxymethyl-lysine- and methylglyoxal (MG)-modified BSA, was assessed in AGER1 overexpressing (AGER1(+) EC) or knockdown (sh-mRNA-AGER1(+) EC) human aortic endothelial (EC) and ECV304 cells, and aortic segments from old (18 mo) C57BL6-F(2) mice, propagated on low-AGE diet (LAGE), or LAGE supplemented with MG (LAGE+MG). Wild-type EC and sh-mRNA-AGER1(+) EC, but not AGER1(+) EC, had high NOX p47(phox) and gp91(phox) activity, superoxide anions, and NF-kappaB p65 nuclear translocation in response to MG and N(epsilon)-carboxymethyl-lysine. These events involved epidermal growth factor receptor-dependent PKC-delta redox-sensitive Tyr-311 and Tyr-332 phosphorylation and were suppressed in AGER1(+) ECs and enhanced in sh-mRNA-AGER1(+) ECs. Aortic ROS, PKC-delta Tyr-311, and Tyr-332 phosphorylation, NOX expression, and nuclear p65 in older LAGE+MG mice were significantly increased above that in age-matched LAGE mice, which had higher levels of AGER1. In conclusion, circulating AGEs induce NADPH-dependent ROS generation in vascular aging in both in vitro and in vivo models. Furthermore, AGER1 provides protection against AGE-induced ROS generation via NADPH. Less
Human aortic endothelial (HAEC) and human coronary artery smooth muscle cell (HCASMC) responses on electrospun silk fibroin scaffolds were studied to evaluate potential f... More
Human aortic endothelial (HAEC) and human coronary artery smooth muscle cell (HCASMC) responses on electrospun silk fibroin scaffolds were studied to evaluate potential for vascular tissue engineering. Cell proliferation studies supported the utility of this biomaterial matrix by both HAECs and HCASMCs. Alignment and elongation of HCASMCs on random non-woven nanofibrous silk scaffolds was observed within 5 days after seeding based on SEM and confocal microscopy. Short cord-like structures formed from HAECs on the scaffolds by day 4, and a complex interconnecting network of capillary tubes with identifiable lumens was demonstrated by day 7. The preservation of cell phenotype on the silk fibroin scaffolds was confirmed by the presence of cell-specific markers, including CD146, VE-cadherin, PECAM-1 and vWF for HAECs, and SM-MHC2 and SM-actin for HCASMCs at both protein and transcription levels using immunocytochemistry and real-time RT-PCR, respectively. Formation of ECM was also demonstrated for the HCASMCs, based on the quantification of collagen type I expression at protein and transcription levels. The results indicate a favorable interaction between vascular cells and electrospun silk fibroin scaffolds. When these results are factored into the useful mechanical properties and slow degradability of this protein biomaterial matrix, potential utility in tissue-engineered blood vessels can be envisioned. Less
Background and objectives: Advanced glycation end products, known pro-inflammatory and pro-oxidative compounds that accumulate in patients with chronic kidney disease, ma... More
Background and objectives: Advanced glycation end products, known pro-inflammatory and pro-oxidative compounds that accumulate in patients with chronic kidney disease, may play a major role in their high prevalence of endothelial dysfunction and subsequent cardiovascular disease. This study examined the association of advanced glycation end product accumulation with cellular receptor for advanced glycation end product expression and endothelial dysfunction as well as the mechanisms of this association in chronic kidney disease. Design, setting, participants, & measurements: A cross-sectional study was conducted of ambulatory patients without diabetes and with different stages of chronic kidney disease (n = 51), compared with gender- and age-matched healthy subjects. Fasting blood was obtained for measurement of advanced glycation end products and mRNA receptor for advanced glycation end product expression in peripheral blood mononuclear cells. Endothelial reactivity was assessed by the microcirculatory response to local ischemia (postocclusive reactive hyperemia) and local hyperthermia (thermal hyperemia). Sera were pooled and passed through affinity columns to separate advanced glycation end product-rich fractions, which were incubated with human aortic endothelial cells, with or without blockade of receptor for advanced glycation end product, to measure their effect on endothelial nitric oxide synthase. Results: Glomerular filtration rate correlated with serum advanced glycation end product, mRNA receptor for advanced glycation end product levels, postocclusive reactive hyperemia, and thermal hyperemia. Serum advanced glycation end product correlated with receptor for advanced glycation end product and inversely with postocclusive reactive hyperemia. Advanced glycation end product-rich fractions from chronic kidney disease sera suppressed endothelial nitric oxide synthase expression of human aortic endothelial cells compared with sera from healthy subjects, an effect abrogated by receptor for advanced glycation end product blockade. Conclusions: This study demonstrates for the first time an association of excess advanced glycation end product burden with increased peripheral blood mononuclear cell mRNA receptor for advanced glycation end product and in vivo endothelial dysfunction in patients with chronic kidney disease. Endothelial dysfunction in chronic kidney disease may be partly mediated by advanced glycation end product-induced inhibition of endothelial nitric oxide synthase through receptor for advanced glycation end product activation. Less
Acute spinal cord injury (SCI) causes progressive hemorrhagic necrosis (PHN), a poorly understood pathological process characterized by hemorrhage and necrosis that leads... More
Acute spinal cord injury (SCI) causes progressive hemorrhagic necrosis (PHN), a poorly understood pathological process characterized by hemorrhage and necrosis that leads to devastating loss of spinal cord tissue, cystic cavitation of the cord, and debilitating neurological dysfunction. Using a rodent model of severe cervical SCI, we tested the hypothesis that sulfonylurea receptor 1-regulated (SUR1-regulated) Ca(2+)-activated, [ATP](i)-sensitive nonspecific cation (NC(Ca-ATP)) channels are involved in PHN. In control rats, SCI caused a progressively expansive lesion with fragmentation of capillaries, hemorrhage that doubled in volume over 12 hours, tissue necrosis, and severe neurological dysfunction. SUR1 expression was upregulated in capillaries and neurons surrounding necrotic lesions. Patch clamp of cultured endothelial cells exposed to hypoxia showed that upregulation of SUR1 was associated with expression of functional SUR1-regulated NC(Ca-ATP) channels. Following SCI, block of SUR1 by glibenclamide or repaglinide or suppression of Abcc8, which encodes for SUR1 by phosphorothioated antisense oligodeoxynucleotide essentially eliminated capillary fragmentation and progressive accumulation of blood, was associated with significant sparing of white matter tracts and a 3-fold reduction in lesion volume, and resulted in marked neurobehavioral functional improvement compared with controls. We conclude that SUR1-regulated NC(Ca-ATP) channels in capillary endothelium are critical to development of PHN and constitute a major target for therapy in SCI. Less
Francisella tularensis, an intracellular pathogen, is highly virulent when inhaled. Alveolar epithelial type I (ATI) and type II (ATII) cells line the majority of the alv... More
Francisella tularensis, an intracellular pathogen, is highly virulent when inhaled. Alveolar epithelial type I (ATI) and type II (ATII) cells line the majority of the alveolar surface and respond to inhaled pathogenic bacteria via cytokine secretion. We hypothesized that these cells contribute to the lung innate immune response to F. tularensis. Results demonstrated that the live vaccine strain (LVS) contacted ATI and ATII cells by 2 h following intranasal inoculation of mice. In culture, primary human ATI or ATII cells, grown on transwell filters, were stimulated on the apical (AP) surface with virulent F. tularensis Schu 4 or LVS. Basolateral (BL) conditioned medium (CM), collected 6 and 24 h later, was added to the BL surfaces of transwell cultures of primary human pulmonary microvasculature endothelial cells (HPMEC) prior to the addition of polymorphonuclear leukocytes (PMNs) or dendritic cells (DCs) to the AP surface. HPMEC responded to S4- or LVS-stimulated ATII, but not ATI, CM as evidenced by PMN and DC migration. Analysis of the AP and BL ATII CM revealed that both F. tularensis strains induced various levels of a variety of cytokines via NF-kappaB activation. ATII cells pretreated with an NF-kappaB inhibitor prior to F. tularensis stimulation substantially decreased interleukin-8 secretion, which did not occur through Toll-like receptor 2, 2/6, 4, or 5 stimulation. These data indicate a crucial role for ATII cells in the innate immune response to F. tularensis. Less